
����̼��ֱ���Ե��ӽṹ��Ӱ��
�����1,2,�� ��1,2,����ɽ2,ŷ����ƽ2
(1.���ϴ�ѧ ������ѧ�뼼��ѧԺ,���� ��ɳ,410083;
2.���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ,���� ��ɳ,410083)
ժ Ҫ��
: �Ա��˵�һ��ԭ���д�ͷ�㷽�����뾭�鷽�����ܶȷ��������ļ������������ĵȲ���,��Ϊ:��armchair�͵���̼�ܽ��а뾭���AM1/STO-3G����ˮƽ�ļ���,�������㹻���ڼ�����϶,��ʡʱ��ѡȡm=n=3,4,5,6,7,8,10�Ƚṹ��armchair��̼��,����AM1�����������ռ�ݷ��ӹ��HOMO����Ϳ�����LUMO�ܼ��Լ�����������ϵͳ����ͷ���,������ʵ����һ�¡��о��������:����armchair������̼��,����϶��ֱ����������³�ָ����˥��,����絼��������;���ͦµ��ӽ���ֲ���̼�ܹܱ���Χ,��������ܶ�i��ֱ�������������̼�����ĵ����ܶ�,�Ҽ��ͦм��������ȼ�С,�侶�����Դ�����,����̼����������������ʯī����,�������仯ѧ���ԡ�
�ؼ���: ����̼��; ��϶; ���ӽṹ
��ͼ�����:TQ018 ���ױ�ʶ��:A ���±��: 1672-7207(2005)01-0044-05
Effect of Diameter on Electronic Structure of Open-ended
Single Wall Carbon Nanotubes
LI Yan-feng1,2, XU Hui1,2, MA Song-shan2, OUYANG Fang-ping2
(1.School of Physics Science and Technology, Central South University, Changsha 410083, China;
2.School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: The computing errors and computing consumptions of ab initio, semi-empirical and density functional transformation (DFT) method were compared with each other. The results show that the Austin Model 1(AM1), which belongs to semi-empirical methods is very fit for the research of energy gap of single wall carbon nanotubes in STO-3G level. Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) energy gap of a series of dimension-variational single wall carbon nanotubes m=n=3,4,5,6,7,8,10 were computed. The results are in agreement with those of experiments. With the increasing of the diameter of tube, the energy gap exponentially reduces and the conductivity increases gradually. �� and �� electrons distribute alternately around the nanotube. With the increase of the diameter of tube, the curvatures of �� and �� bonds reduce, the electron density closes up the surface of tube, and the radial character and chemical activity of the nanotube reduce.
Key words: single wall carbon nanotube; energy gap; electronic structure
�Դ�S.IIJIMA�Ʊ�������̼������[1],����̼�������ڷ����������ϲ�������������DZ��Ӧ�ü�ֵ����Ϊ���������ϡ���ѧ����о��ȵ�[2-5],̼�ܵĵ��ӽṹ�ͼ�������ֱ�Ӿ��������ڲ���Ӧ���е�����,��������뾶�������Ȳ�ͬ,����̼�ܿ��Գ��ֽ�����뵼����Ե�������[6-8]��
һЩ�о���Ӧ������ѧԭ�����������˽ṹ��̼�����ܵ�Ӱ�����[9,10]��J.W.WINTMIRE���õ�һ��ԭ��LDT����������zigzag�ܵ���϶��뾶�Ĺ�ϵ,�������ڰ뾶�仯1 nm����,���õ��뾶�仯�ϴ�ʱ̼�ܵ�ʵ�ʱ仯����[11]��L.TUKER���о���zigzag�ܵ������ռ�ݷ��ӹ��(HOMO)����Ϳ�����(LUMO)�ֲ���뾶�Ĺ�ϵ[12]��̼�ܵĽṹ��Ӱ��̼�����ܵĹؼ����ء�Ϊ��,�������ݵ�һԭ���еİ뾭�鷽��AM1(Austin Model 1),��һϵ�в�ֱͬ����armchair�͵���̼�ܵ��ӽṹ���м���,��������Ϻ���϶������
1 ���㷽����ԭ��
�뾭����Ƽ���ķ�����2����ͬ��·��:һ���Ƕ�������ܵĻ��ֽ��м���,ѡ��һЩ�������������,������һЩ��,���ݿ˶����ӹ����HMO��;��һ��;����ֱ�ӻ�����ѧ��ʽ,��������ܾ���Ԫ�г��ֵ�ԭ�Ӻͷ��ӻ���,����һЩ���ơ����ڽ��ƴ���������ͬ,����ó���һЩ[CM(22] ��ͬ�ķ���,��ȫ�����ص���(CNDO)��������[CM)][] �ص���(INDO)�ͺ���˫ԭ�����ص���(NDDO)�ȡ�
AM1�ǻ���NDDO���Ƶİ뾭�������ѧ���㷽��,ͨ������Hartree-Fock(HF)������˫ԭ�ӻ���![]() ���ص���,�����Ż��IJ����������[13]��
���ص���,�����Ż��IJ����������[13]��
ab initio-HF���� ��������˫ԭ���ص���(Modified Neglect of Diatomic Overlap, MNDO)�� �뾭�鷽��AM1���ܶȷ�������(DFT)�ļ����� �����ģ�Ȳ���[14]�Ա����1��ʾ�� �ɼ�, ���ô�ͷ����ܶȷ�������õ��Ľ�����ȸ�, ��ʵ����, ���ѡȡ�ϴ�Ļ���, �������ĵ�CPUʱ�䡢 �ڴ����Դ��������ӡ�
���������о����ص�����϶�����Ժ�ֱ���仯�Ĺ�����,�Լ��㾫��Ҫ��,����Ҫ�����ģ�ͽ϶�,����,���ð뾭�鷨AM1�õ��Ľ���㹻�����о���϶���ɡ�����,���ð뾭�鷨AM1�����ܼ�����,��ʹ��Slater��STO-3G������顣
Ŀǰ,��ʵ���ҶԵ���̼�ܵ��Ʊ��������൱����[15-19],���о��Ե��ڹ�armchair��Ϊ��,ϵͳ�������̼�ܵ���϶�ṹ���ɡ�
̼�ܵļ��ι���Ϊ:h=na1+ma2������:a1��a2Ϊ��λʸ������m=n,��=30��ʱ,̼�ܼ���ģ��Ϊarmchairģ�͡�T.SATO�ȵ��о��������,̼����϶��ֱ����������˥��[20],����,Ϊ��������뾶�仯ʱ���ȵ�Ӱ��,����ʱ�����þ���12��̼��Ĺܽṹ(n=3����),����̼�ܾ�����Hԭ�ӱ��Ͷ˿�Cԭ�ӵ����Ҽ�[21]��
�������õ�ģ����ͼ1��ʾ��
�� 1 ��ͷ�㡢�뾭����ܶȷ����������ܶԱ�
Table 1 Comparison of ab initio, semi-empirical and DFT Method 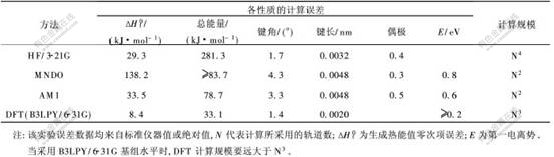

m=n: (a)��3; (b)��4; (c)��5; (d)��6; (e)��8; (f)��10
ͼ 1 armchair��̼�ܽṹͼ
Fig. 1 Geometrical structure of single wall carbon nanotubes
2 ��������
���ڴ���̼��,��϶�͵��ӽṹֱ��Ӱ����������ѧ����,ͨ���о���϶�ͼ�������,�ɵõ�̼����Ӧ���ϵ���Ϣ���ֱ���������̼�������ռ�ݹ��������ֵEHOMO�����δռ�ݹ��������ֵELUMO,������:
��Eg=ELUMO-EHOMO��(1)
����:��EgΪ��϶,��ֵ��ֱ�ӷ�ӳ̼�ܵĻ�ѧ���ԡ�̼����϶ֵ���2��ʾ��
����������,armchair��̼����϶��̼��ֱ����������³�ָ���ݼ�,���л��в��ֳ���С����˥��������������������T.W.ODOW�ȵ�ʵ������һ��[3]��������Ϊ,���ð뾭���AM1����,��������ȡ����ա�̼��ģ��,���о�����̼�ܹܾ��仯����϶�����ӽṹ�����Ǻ��ʵġ�̼��ֱ��Ϊ[6]:
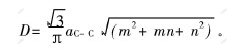
����:aC-CΪ̼̼������̼��ֱ��-��϶ͼ��ͼ2��ʾ����ͼ2�ɼ�,����ֱ��������,̼�ܵ���϶Ѹ�ټ�С��������Ϊ̼��ֱ������,������Խ��ԽС,�ӽ�����ʯī��ṹ,��ܽṹӰ���С,��ṹӰ������,������絼������,̼�ܽ������ɷǽ�����������͵�ת�����ӱ�2��֪,����̼�ܵķ����ܼ�Ҳ��̼��ֱ�����������,���,����Ϊ�仯ѧ������̼�ܹܾ���������С��
�� 2 ̼����϶ֵ
Table 2 Data of energy gap of carbon nanotube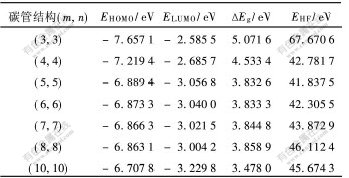
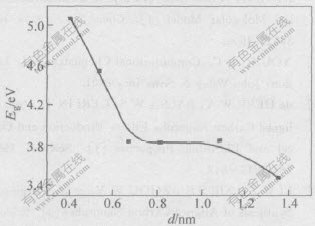
ͼ 2 armchair��̼����϶(Eg)��ֱ���Ĺ�ϵ
Fig. 2 Relationship between energy gap and
diameter of armchair carbon nanotube
ͨ���о�������,����̼���е�C��C����ҪΪ��Cԭ�ӵ�2s��2p�۵����ӻ����ɵĦҼ��ͦм�,ÿ��Cԭ�Ӿ�����Χ��Cԭ�Ӳ��æҼ����;ͬʱ,�м�����̼�ܱ�����,���Լ���������,�е�������̼�ܱ��������˶�,����HOMO��LUMO��Ϊ���͵Ħ��ͷ����ͦ��ͳɼ����,����,̼�ܵĵ絼������Ҫ�Цм�������
n=3,4,5,10ʱ̼�ܵ��ܵ����ܶȷֲ���n=7ʱ�ķ��ӹ��������ͼ3��ͼ4��ʾ��
��ͼ4�ɼ�,�����Ӻͦµ��ӽ���ֲ���̼�ܵ�����,���ؾ�������Ӻͦµ���Ҳ�ʽ���ֲ�������ͼ3���Կ���,������ϳ̶���С,�����ܶ�����������̼�ܱ�,�侶��絼������,����絼����,���ڵ���ʯī��絼ֵ,��絼����ԭ���ķǽ������ɵ������͡���ֱ��D������ʱ,�м���������С,����������ʯī�IJ�ṹ,ԭ���������ͽṹӰ����С,������������С,�����������,����ʯīƬ�����۽���ܡ�
�ɴ˿ɼ�,����̼�ܾ����ȶ��IJ��Һܸߵ�ǿ�Ⱥն�,�������ڸ��ϲ����з�����ģ�����á�
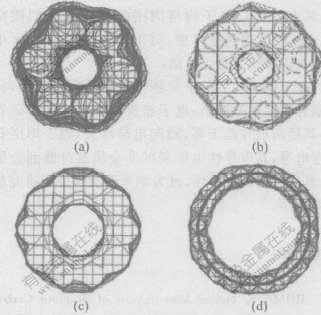
(a)��n=3; (b)n=4; (c)n=5; (d)n=10
ͼ 3 ̼���ܵ����ܶ�ͼ(����۲�)
Fig. 3 Total electronic density of carbon
nanotubes ( axis direction)

����������; �����µ���
ͼ 4 n=7ʱarmchair������̼�ܷ��ӹ������ͼ
Fig. 4 Surface of molecular orbit of single
wall carbon nanotube when n=7
3 �� ��
a. ���õ�һԭ���е�AM1�뾭�鷨������һϵ��ֱ���仯��armchair������̼�ܵ��ܼ��͵��ӽṹ,����̼�ܵ���϶��Eg��ֱ���������ָ��������˥��,����̼�ܵķ����ܼ�Ҳ��ֱ�����������,�仯ѧ������ܾ���������С��
b. ����̼�ܵļ�����ʯī��ṹ�ļ�������,������2s��2p����۵����ӻ����ɵĦҼ��ͦм�,����ÿ��Cԭ�Ӿ�����Χ��Cԭ�Ӳ��ü����,�м�����̼�ܱ�����,�䲻�Լ���������,�е�������̼�ܱ��������˶�,
c. �����Ӻͦµ��ӽ���ֲ���̼�ܵ�����,������ϳ̶���С,�����ܶ�����������̼�ܱ�,�侶��絼������,����絼����,���ڵ���ʯī��絼,��絼����ԭ���ķǽ������ɵ�������,����һ���Ĺ�����,��Ϊ����̼�ܵ�Ӧ���о���ָ�����á�
�����:
[1]IIJIMA S. Helical Microtubules of Graphitic Carbon[J]. Nature, 1991, 354: 56-58.
[2]WILDOER J W G, VENEMA L C, RINZLER A G. Electronic Structure of Atomically Resolved Carbon Nanotubes[J]. Nature, 1998, 391:59-61.
[3]ODOM T W, HUANG J L, KIM P, et al. Atomic Structure and Electronic Properties of Single-walled Carbon Nanotubes[J]. Nature, 1998, 391: 62-64.
[4]TSUKAGOSHI K, YONEYA N, URYU S, et al. Carbon Nanotube Devices for Nanoelectronics[J]. Phys B, 2002, 323: 107-114.
[5]DAI Hong-jie. Carbon Nanotubes: Opportunities and Challenges[J]. Surface Science , 2002, 500(1-3): 218-241.
[6]������,IJ����. ���ײ��Ϻ����ṹ[M]. ����:��ѧ������,2001.
ZHANG Li-de, MOU Ji-mei. Nano Material and Nano Structure [M]. Beijing: Science Press, 2001.
[7]LANGER L, STOCKMAN L, HEREMANS J P, et al. Electrical Resistance of a Carbon Nanotube Bundle[J]. J Mater Res, 1994,9: 927-932.
[8]SAITE R, DRESSELHAUS G, DRESSELHAUS M S. Electronic Structure of Double-layer Grapheme Tubules[J]. J Appl Phys, 1993, 73(2):494-500.
[9]SAITO R, FUJITA M. Electronic Structure of Chiral Grapheme Tubules[J]. Appl Phys Lett, 1992, 60(18): 2204-2209.
[10]HAMADA N, SAWADA S, OSHIYAMA A. New One-dimensional Conductors: Graphitic Microtubules[J]. Phys Rev Lett, 1992, 68(10): 1579-1581.
[11]MINTMIRE J W , WHITE C T. Electronic and Structural Properties of Carbon Nanotube[J]. Carbon, 1995, 33(7):893-902.
[12]TUKER L, ERKOC S. Electronic Properties of Open-ended Single Wall Carbon Nanotubes [J]. Journal of Molecular Structure: THEOCHEM, 2002, 577(2-3): 131-135.
[13]DEWAR M J S, ZOEBISCH E G , HEALY E F, et al. AM1: A new General Purpose Quantum Mechanical Molecular Model[J]. Chem Soc, 1995, 107: 3902-3909.
[14]YOUNG D C. Computational Chemistry[M]. London: John Wiley & Sons Inc, 2001.
[15]de HEER W A, BACSA W S, GERFIN T, et al. Aligned Carbon Nanotube Films: Production and Optical and Electronic Properties[J]. Science, 1995, 268: 845-848.
[16]LI W Z , XIE S S , ZHOU W Y, et al. Large-scale Synthesis of Aligned Carbon Nanotubes[J]. Science, 1996, 274: 1701-1705.
[17]BACHTOLD A, STRUNK C, SALVETAT J P, et al. Aharonov-Bohm Oscillations in Carbon Nanotube[J]. Nature, 1999, 397:673-675.
[18]COLLINS P G, BRADLEY K, ISHIGAMI M, et al. Extreme Oxygen Sensitivity of Electronic Properties of Carbon Nanotube[J]. Science, 2000, 287: 1801-1804.
[19]IIJIMA S. Carbon Nanotubes: Past, Present and Future[J]. Phys B, 2002, 323(1-4): 1-5.
[20]SATO T, TANAKA M, YAMABE T. Size-dependent HOMO-LUMO Gap Oscillation of Carbon Nanotube with a Finite Length[J]. Synthetic Metals, 1999, 103(1-3): 2525-2526.
[21]CASANAS J, ILLAS F, SANZ F, et al. Dissociative Chemisorption of Molecular Hydrogen on Graphite: A MINDO/3 Study[J]. Surf Sci, 1983,133:29-37.
�ո�����:2004-10-26
������Ŀ:���ҽ�������ʿ�����������Ŀ(20020533001)
�����:�����(1979- ),��,����֣����,˶ʿ,���²������Լ�����о�
������ϵ��: �����,��,˶ʿ;E-mail: liyanfeng@qgzxol.com
ժҪ: �Ա��˵�һ��ԭ���д�ͷ�㷽�����뾭�鷽�����ܶȷ��������ļ������������ĵȲ���,��Ϊ:��armchair�͵���̼�ܽ��а뾭���AM1/STO-3G����ˮƽ�ļ���,�������㹻���ڼ�����϶,��ʡʱ��ѡȡm=n=3,4,5,6,7,8,10�Ƚṹ��armchair��̼��,����AM1�����������ռ�ݷ��ӹ��HOMO����Ϳ�����LUMO�ܼ��Լ�����������ϵͳ����ͷ���,������ʵ����һ�¡��о��������:����armchair������̼��,����϶��ֱ����������³�ָ����˥��,����絼��������;���ͦµ��ӽ���ֲ���̼�ܹܱ���Χ,��������ܶ�i��ֱ�������������̼�����ĵ����ܶ�,�Ҽ��ͦм��������ȼ�С,�侶�����Դ�����,����̼����������������ʯī����,�������仯ѧ���ԡ�
�ؼ���: ����̼��; ��϶; ���ӽṹ
��ͼ�����:TQ018 ���ױ�ʶ��:A ���±��: 1672-7207(2005)01-0044-05
