

A first-principle calculation of structural, mechanical and electronic properties of titanium borides
YAN Hai-yan1, WEI Qun2, CHANG Shao-mei3, GUO Ping4
1. Department of Chemistry and Chemical Engineering, Baoji University of Arts and Sciences, Baoji 721013, China;
2. Department of Physics, Xidian University, Xi��an 710071, China;
3. Department of Physics, Baoji University of Arts and Sciences, Baoji 721007, China;
4. Department of Physics, Northwest University, Xi��an 710069, China
Received 17 August 2010; accepted 15 November 2010
Abstract:
The first-principle calculations are performed to investigate the structural, mechanical and electronic properties of titanium borides (Ti2B, TiB and TiB2). Those calculated lattice parameters are in good agreement with the experimental data and previous theoretical values. All these borides are found to be mechanically stable at ambient pressure. Compared with parent metal Ti (120 GPa), the larger bulk modulus of these borides increase successively with the increase of the boron content in three borides, which may be due to direction bonding introduced by the boron atoms in the lattice and the strong covalent Ti��B bonds. Additionally, TiB can be regarded as a candidate of incompressible and hard material besides TiB2. Furthermore, the elastic anisotropy and Debye temperatures are also discussed by investigating the elastic constants and moduli. Electronic density of states and atomic Mulliken charges analysis show that chemical bonding in these titanium borides is a complex mixture of covalent, ionic, and metallic characters.
Key words:
titanium borides; first-principle calculations; mechanical properties; density of states; Mulliken atomic population analysis;
1 Introduction
With the actual increasing interest and impetus in the search for materials possessing specific and desired properties, transition metal borides have attracted much attention for their fundamental physics and technological application [1-3]. More precisely, titanium borides (Ti2B, TiB and TiB2) are a group of materials with outstanding properties such as chemical stability, high hardness, high melting point, good thermal conductivity, and low electrical resistivity [4-5]. Besides bulk titanium borides, increasing attention is also devoted recently to their nano-sized forms such as nanopowders, nanowires, nanotubes, and nanocomposites [6-8].
Among these borides, the most known AlB2-type TiB2 is a potential candidate for many high-performance applications, in cutting tools, wear-resistant coating, electrodes in aluminum-extraction cells, etc. A great deal of experimental and theoretical effort has been performed on TiB2 at ambient pressure as well as high pressure [9-18]. However, some previous experimental results for the measured mechanical properties are somewhat different [10, 13, 19]. Besides, by contrast, it turned out that the works (including ab initio calculations) for other borides (especially Ti2B) are limited. So, this is one of motivations for the present work. Furthermore, to the best of our knowledge, TiB whiskers as reinforcements play an important role in various titanium alloys [20], while Ti2B is an essentially intermetallic compound [21]. Therefore, systematic studies on the crystal structure and related physical properties of titanium borides are necessary and of great interest.
In the present work, in order to get insight to the mechanical and electronic properties of transition borides systematically, a first-principle study for Ti2B, TiB and TiB2 has been performed using the projector augmented wave (PAW) method. We have evaluated and analyzed a set of physical parameters of these transition borides such as lattice parameters, density of states, Mulliken charges, and mechanical properties (elastic constants, bulk and shear modulus, elastic modulus, elastic anisotropy, etc.). Additionally, the numerical estimates of sound velocities and Debye temperature of three borides were also predicted.
2 Computational methods
The first-principle calculations were performed with the plane-wave basis based on the density functional theory, as implemented in the Vienna ab initio simulation package [22]. The electron and core interactions were included by using the frozen-core all-electron projector augmented wave (PAW) method [23]. The exchange correlation function was treated by the generalized gradient approximation (GGA) proposed by PERDEW, BURKE and ERNZERHOF (PBE) [24]. The plane-wave basis set cut-off was set as 420 eV for all cases. The special points sampling integration over the Brillouin zone was employed by using the Monkhorst-Pack scheme [25] with a grid of 0.035 ?-1. Convergence of the total energy with different number of k points and the plane-wave cut-off energy has been tested. The tolerances for geometry optimization were set as the difference in total energy within 1��10-6 meV/atom. The elastic constants were determined by applying a set of given homogeneous deformation with a finite value and calculating the resulting stress with respect to optimizing the internal atomic freedoms, as implemented by MILMAN and WARREN [26]. Atoms were allowed to relax until Hellman�CFeynman forces were less than 0.001 eV/? and the maximum strain value was 0.2%. The bulk modulus, shear modulus, elastic modulus, and Poisson ratio were thus derived from the Voigt-Reuss- Hill approximation [27].
3 Results and discussion
3.1 Crystal structure parameters
Almost all known transition metal semi-borides crystallize in a body centered tetragonal (Al2Cu-type, Z=4) structure (D4h18) with space group I4/mcm [21]. Hence, for Ti2B, there are four formula units (f. u.) in the conventional tetragonal cell (Fig. 1(a)), in which the B and Ti atoms occupy the 4a (0, 0, 0) and 6h (1/6, 2/3, 0) positions, respectively. There exists an ambiguity regarding the crystal structure of TiB. WYCKOFF [28] reported TiB in FeB structure, whereas PEARSON [29] and LANDOLT-B?RNSTEIN [30] suggested NaCl and ZrS structure, respectively. However, recent investigations [31] have confirmed the FeB configuration. In our calculations, we have taken the orthorhombic FeB structure with the unit cell containing four f. u. (Fig. 1(b)). TiB2 typically crystallizes in the well-known AlB2-type structure (P6/mmm, Z=1) with Ti and B atoms sitting at the origin and (1/3, 2/3, 1/2) sites (Fig. 1(c)), in which the coplanar graphite-like B layers are present alternatively with the close-packed Ti sheets.
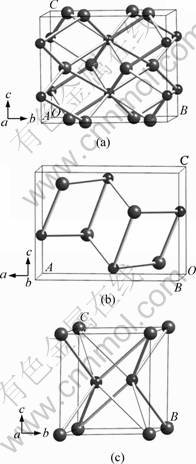
Fig. 1 Crystal structure of Ti2B (a), TiB (b) and TiB2 (c)) (Large and small spheres represent Ti and B atoms, respectively; a, b, c, A, B and C represent directions of crystal cells)
For each compound, the crystal structure was fully optimized with respect to the total energy to determine the lattice parameters: a, b, and c. The calculated lattice constants are given in Table 1, together with the available experimental data and previous theoretical values for comparison [9, 12, 14-17, 21, 31-34]. As seen from Table 1, agreement between the results of the present calculations and experimental data is very good: the deviations between the experimental and theoretical values for TiB and TiB2 are all less than 1%. For tetragonal Ti2B, the calculated lattice parameter a/c is 5.647/4.741, which is in excellent agreement with those previous theoretical calculations. However, compared with the experimental results [21], the highest disagreement in lattice constant a (7%) is obtained for Ti2B. Here we shall emphasize that the transitional metal borides with metal-deficient or boron-deficient compositions can be easily obtained in actual experiments [35]. In one word, the agreement of the structural to be published results confirms the accuracy and reliability of the computational procedure employed.
Table 1 Calculated equilibrium lattice parameters a, b, c, and volume V for Ti2B, TiB, and TiB2 compounds
3.2 Mechanical properties
The accurate calculation of elasticity is essential for understanding the macroscopic mechanical properties of solid. Elasticity describes the response of a crystal under external strain and provides key information of the strength of the material, as characterized by elastic constants. To our knowledge, there is almost no experimental data about the elastic constants for Ti2B and TiB except for TiB2. We hope that our work could provide a useful reference for future study. The complete sets of zero-pressure single crystal elastic constants were calculated by using strain��stress method, as shown in Table 2. The elastic stability is a necessary condition for a stable crystal. A tetragonal crystal has to obey the following restrictions of its elastic constants [36]: C11>0, C33>0, C44>0, C66>0, (C11-C12)>0, (C11+C33-2C13)>0 and 2(C11+C12)+C33+4C13>0. For a stable orthorhombic structure, Cij has to satisfy the elastic stability criteria: C11>0, C22>0, C33>0, C44>0, C55>0, C66>0, C11+C22+C33+ 2(C12+C13+C23)>0, (C11+C22-2C12)>0, (C11+C33-2C13)>0 and (C22+C33-2C23)>0. For the hexagonal structure with five independent elastic constants C11, C33, C44, C12 and C13, they are C33>0, C44>0, C11>|C12|, (C11+2C12)C33> 2C132. In terms of the above conditions, the calculated elastic constants (Table 2) indicate that all these borides are mechanically stable. Our calculated elastic constants for TiB2 are consistent with experimental and previous theoretical results [10-11, 13-14, 16, 19, 33, 37-38]. In addition, one should be aware that the values presented in Table 2 were obtained at T=0 K, and that temperature effects generally reduce the elastic constants. Therefore, we expect the experimental data for Ti2B and TiB at room temperature to be smaller than those listed here.
Using the calculated elastic constants Cij, bulk modulus and shear modulus for the corresponding polycrystalline aggregate are thus determined by the Voig-Reuss-Hill approximation method [27]. Furthermore, the elastic modulus E and Poisson ratio v are obtained in the light of the following equations:
![]() (1)
(1)
![]() (2)
(2)
The calculated bulk modulus, shear modulus, elastic modulus and Poisson ratio are listed in Table 2. Clearly, the calculated results for TiB2 agree well with corresponding experiment [11], supporting the accuracy and reliability of our elastic calculations. For TiB, the large bulk modulus (218 GPa) manifests its strong incompressibility besides TiB2. As expected, all these borides have larger bulk modulus than their parent metal (120 GPa) due to the direction bonding introduced by the boron atoms in the lattice. Moreover, it can be seen that the bulk modulus increases from 173 GPa to 262 GPa with the increase of the boron content in three borides, which are mainly due to the fact that the atomic volume decreases with the increase in the boron content in these borides. Furthermore, as a better indicator of potential hardness for materials, shear modulus quantifies the resistance to the shear deformation. Compared with the well-known hard TiB2, TiB has a relatively large shear modulus shown in Table 2, indicating that it is expected to withstand shear strain to a certain extent and may act as hard material. By contrast, Ti2B possesses much lower shear modulus than TiB and TiB2, resulting in low bond-bending force constants and consequently a low hardness.
Table 2 Calculated elastic constants Cij, bulk modulus B, shear modulus G, elastic modulus E and Poisson ratio v for three borides
It is well known that microcracks are easily induced in the materials due to the significant elastic anisotropy. Therefore, it is important to calculate elastic anisotropy in order to improve its mechanical durability. In view of the high bulk and shear moduli of TiB and TiB2, we here have calculated the bulk modulus B along the different crystallographic axes for these two compounds. The anisotropy of the bulk modulus along the a-axis and b-axis with respect to the c-axis are given by ABa=Ba/Bc and ABb=Bb/Bc, where Ba, Bb, and Bc are the bulk modulus along different crystal axes, defined as Bi=idP/di, i=a, b, and c. Note that a value of 1.0 indicates elastic isotropy and any deviation from 1.0 represents elastic anisotropy. Using the relations mentioned above, the calculated parameters about elastic anisotropy together with the available experimental data are shown in Table 3. From our calculations, it is clear that both compounds are all elastic anisotropic. For TiB, it is interesting to note that the directional bulk modulus along the b-axis is larger than that along the a-axis or c-axis, which is consistent with the predicted elastic constants along different axes (see Table 2). Compared with TiB, TiB2 is more anisotropic characterized by larger anisotropy of the bulk modulus ABa and ABb, which are also in agreement with experimental values [11].
As a fundamental parameter, Debye temperature closely relates to many physical properties of solids, such as specific heat, dynamic properties, and melting temperature [39]. At low temperature, it can be calculated from the elastic constants using the average sound velocity vm, by the following equation:
![]() (3)
(3)
where h is Planck��s constant; k is Boltzmann��s constant; NA is Avogadro��s number; n is the number of atoms per formula unit; M is the molecular mass per formula unit; �� is the density. The average sound velocity vm is given by
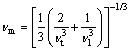 (4)
(4)
where vt and vl are the transverse and longitudinal elastic wave velocities of the polycrystalline materials, respectively, and are given by Navier��s equation [40]. The calculated longitudinal, transverse and average elastic velocities, and Debye temperatures for three titanium borides are listed in Table 4. The calculated Debye temperature for TiB2 is higher than that of Ti2B or TiB, which agrees well with the experimental results [11, 41].
3.3 Electronic properties
To understand the mechanical properties of these borides on a fundamental level, the site projected and total electronic densities of states (DOS) of three titanium borides were calculated at zero pressure, as plotted in Fig. 2. It can be seen that all these compounds possess finite DOS at the Fermi level. Hence, these borides exhibit clear metallic behavior in their crystalline state. Moreover, the major orbital occupancy near Fermi level stems is predominated by the Ti-3d electrons, which are the principal cause for their metallicity. In Fig. 2, the DOS of Ti-3d and B-2p are energetically degenerated from the bottom of valence band to the Fermi level, indicating the covalent hybridization between the Ti and B atoms in these compounds, especially for TiB2. It is earlier proposed that the lower DOS at the Fermi level often characterizes a more stable structure [42-43]. Among three borides, TiB2 possesses the lowest DOS on the Fermi level, indicating its highest stability. Moreover, the decrease in the total DOS at the Fermi level is accompanied with the appearance of the pseudo gap around the Fermi level (Fig. 2(c)), which is helpful to increase the stability of TiB2 and the strength of the covalent of the chemical bond between the Ti and B atoms. Since the coincidence between the pseudogap and the Fermi level in the TiB2 is due to the band-filling effect and depends only on the electron number, it must be also noted that the presence of the Fermi level near the minimum of the DOS histogram indicates that the bonding states are occupied when all the antibonding ones are empty. So TiB2 has better cohesively than Ti2B and TiB. This is confirmed through the large bulk modulus of TiB2 compared with Ti2B and TiB.
Table 3 Calculated directional bulk modulus Ba, Bb, Bc, anisotropy factors ABa, and ABb of TiB and TiB2
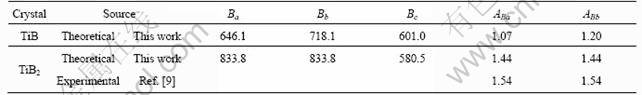
Table 4 Longitudinal, transverse, average elastic wave velocities (vl, vt and vm), and Debye temperature ��D at theoretical equilibrium volume


Fig. 2 Calculated total and partial state density of Ti2B (a), TiB (b) and TiB2 (c)
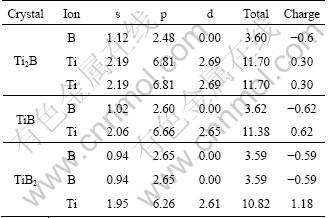
In order to understand the changes of the DOS on the Fermi level, we calculated the charge transfer situation between Ti and B atoms of Ti2B, TiB and TiB2 by the Mulliken atomic population analysis and tabulated them in Table 5. In Table 5, Ti atoms of TiB2 have the largest charge values among the three titanium borides. The large charge transfering from Ti to B atoms of TiB2 mainly comes from more electrons of Ti-3d orbital of TiB2 taking part in the covalent bonding between Ti and B atoms or transferring from Ti and B atoms, which can be seen from the smallest Mulliken charge value (2.61). Consequently, the more Ti-3d electrons of TiB2 become the bonding states than Ti2B and TiB, which is accompanied with the larger decrease of Ti-3d states on the Fermi level. In addition, we found a charge transferring from Ti to B atom for all these borides, implying an ionic contribution to Ti��B bonding. We thus conclude that the chemical bonding in these titanium borides is a complex mixture of covalent, ionic and metallic characters. Such a conclusion was also found in other transition metal borides [15, 44-45].
Table 5 Calculated atomic population of three titanium borides
4 Conclusions
1) Our calculated lattice parameters are in good agreement with the experimental data and previous theoretical values for these titanium borides. The calculated elastic constants indicate that all these borides are mechanically stable at ambient pressure.
2) The larger bulk modulus of these borides increase successively from Ti2B to TiB2 compared with parent metal Ti, which may be due to direction bonding introduced by the boron atoms in the lattice and the strong covalent Ti-B bonds. Additionally, TiB and TiB2 show different degrees of elastic anisotropy. Debye temperatures calculated by elastic constants are 648 K for Ti2B, 967.7 K for TiB, and 1 204.5 K for TiB2.
3) According to the electronic density of states and Mulliken atomic population analysis, we thus conclude that the chemical bonding in these titanium borides is a complex mixture of covalent, ionic, and metallic characters
References
[1] LUNDSTORM T, RUNDQVIST S. Borides, silicides and phosphides [M]. London: Methuen, 1965.
[2] Serebriakova T I, Neronov V A, Peshev P D. High-temperature borides [M]. Moscow: Metallurgy, 1991. (in Russian)
[3] Basu B, Raju G B, Suri A K. Processing and properties of monolithic TiB2 based materials [J]. Int Mater Rev, 2006, 51(6): 352-374.
[4] Tian D C, Wang X B. Electronic structure and equation of state of TiB2 [J]. J Phys: Condens Matter, 1992, 4(45): 8765-8772.
[5] Atri R R, Ravichandran K S, Jha S K. Elastic properties of in-situ processed Ti-TiB composites measured by impulse excitation of vibration [J]. Mater Sci Eng A, 1999, 271(1-2): 150-159.
[6] Mitterer C, Losbichler P, Hofer F, Warbichler P, PGibson N, Gissler W. Nanocrystalline hard coatings within the quasi-binary system TiN-TiB2 [J]. Vacuum, 1998, 50(3-4): 313-318.
[7] Mishra S K, Rupa P K P, Pathak L C. Surface and nanoindentation studies on nanocrystalline titanium diboride thin film deposited by magnetron sputtering [J]. Thin Solid Films, 2007, 515(17): 6884-6889.
[8] Kartal G, Timur S, Urgen M, Erdemir A. Electrochemical boriding of titanium for improved mechanical properties [J]. Surf Coat Technol, 2010, 204(23): 3935-3939.
[9] Post B, WGlaser F, Moskowitz D. Transition metal diborides [J]. Acta Metall, 1954, 2(1): 20-25.
[10] Gilman J J, Roberts B W. Elastic constants of TiC and TiB2 [J]. J Appl Phys, 1961, 32(7): 1405.
[11] Spoor P S, Maynard J D, Pan M J, Green D J, Hellmann J R, Tanaka T. Elastic constants and crystal anisotropy of titanium diboride [J]. Appl Phys Lett, 1997, 70(15): 1959-1961.
[12] Munroe R G. Material properties of titanium diboride [J]. J Res Natl Inst Stand Technol, 2000, 105(5): 709-720.
[13] Manghnani M H, Fisher E S, Li F Y, Grady D E. Elastic moduli of TiB2 [J]. Ceram Trans, 1993, 38(4): 771-785.
[14] Milman V, Warren M C. Elastic properties of TiB2 and MgB2 [J]. J Phys: Condens Matter, 2001, 13(24): 5585-5595.
[15] Vajeeston P, Ravindran P, Ravi C, Asokamani R. Electronic structure, bonding, and ground-state properties of AlB2-type transition-metal diborides [J]. Phys Rev B, 2001, 63(4): 045115.
[16] Perottoni C A, Pereira A S, Jornada J A H da. Periodic Hartree-Fock linear combination of crystalline orbitals calculation of the structure, equation of state and elastic properties of titanium diboride [J]. J Phys: Condens Matter, 2000, 12(32): 7205-7222.
[17] Campa P E V, Dorena V E V. Ground state properties of titaniumdiboride [J]. High Pressure Research, 1995, 13(6): 335-341.
[18] Peng F, Fu H Z, Cheng X L. First-principles calculations of thermodynamic properties of TiB2 at high pressure [J]. Physica B, 2007, 400(1-2): 83-87.
[19] Wright S. Estimation of single-crystal elastic constants from textured polycrystal measurements [J]. J Appl Crystallogr, 1994, 27(5): 794-801.
[20] Feng H B, Zhou Y, Jia D C, Meng Q C, Rao J C. Growth mechanism of in situ TiB whiskers in spark plasma sintered TiB/Ti metal matrix composites [J]. Crystal Growth & Design, 2006, 6(7): 1626-1630.
[21] Mohn P. The calculated electronic and magnetic properties of the tetragonal transition-metal semi-borides [J]. J Phys C: Solid State Phys, 1988, 21(15): 2841-2851.
[22] Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Phys Rev B, 1999, 59(3): 1758-1775.
[23] Bl?uml P E. Projector augmented-wave method [J]. Phys Rev B, 1994, 50(24): 17953-17979.
[24] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple [J]. Phys Rev Lett, 1996, 77(18): 3865-3868.
[25] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations [J]. Phys Rev B, 1976, 13(12): 5188-5192.
[26] Milman V, Warren M C. Elasticity of hexagonal BeO [J]. J Phys: Condens Matter, 2001, 13(2): 241-251.
[27] Hill R. On discontinuous plastic states, with special reference to localized necking in thin sheets [J]. J Mech Phys Solids, 1952, 1(1): 19-30.
[28] Wyckoff W G. Crystal structures [M]. New York: Wiley, 1963.
[29] Pearson W P. Handbook of lattice spacings and structures of metals and alloys [M]. New York: Pergamon, 1967.
[30] Landolt-B?rnstein. Numerical data and functional relationships in science and technology [M]. Berlin: Springer, 1971.
[31] Mohn P, Pettifor D G. The calculated electronic and structural properties of the transition-metal monoborides [J]. J Phys C: Solid State Phys, 1988, 21(15): 2829-2839.
[32] Mouffok B, Feraoun H, Aourag H. Electronic structure of some mono-, semi-titanium boride and diboride [J]. Mater Lett, 2006, 60(12): 1433-1436.
[33] Panda K B, Chandran K S R. First principles determination of elastic constants and chemical bonding of titanium boride (TiB) on the basis of density functional theory [J]. Acta Mater, 2006: 541641-541657.
[34] Arnold H S, Toshimoto K. Nuclear magnetic resonance in transition-metal diborides [J]. J Chem Phys, 1963, 38(4): 865-872.
[35] Hao X F, Xu Y H, Wu Z J, Zhou D F, Liu X J, Cao X Q. Low-compressibility and hard materials ReB2 and WB2: Prediction from first-principles study [J]. Phys Rev B, 2006, 74(22): 224112.
[36] Wu Z J, Zhao E J, Xiang H P, Hao X F, Liu X J, Meng J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles [J]. Phys Rev B, 2007, 76(5): 054115.
[37] YAO Qiang, XING Hui, MENG Li-jun, SUN Jian. Theoretical calculation of elastic properties of TiB2 and TiB [J]. Chinese Journal of Nonferrous Metals, 2007, 17(8): 1297-1301. (in Chinese)
[38] Panda. K B, Chandran K S R. Determination of elastic constants of titanium diboride (TiB2) from first principles using FLAPW implementation of the density functional theory [J]. Comput Mater Sci, 2006, 35(2): 134-150.
[39] Ravindran P, Fast L, Korzhavyi P A, Johansson B, Wills J, Eriksson O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2 [J]. J Appl Phys, 1998, 84(9): 4891-4904.
[40] Schreiber E, Anderson O L, Soga N. Elastic constants and their measurements [M]. New York: McGraw-Hill, 1973.
[41] Ledbetter H, Tanaka T. Elastic-stiffness coefficients of titanium diboride [J]. J Res Nat Inst Stand Technol, 2009, 114(6): 333-339.
[42] Xu J H, Freeman A. Phase stability and electronic structure of ScAl3 and ZrAl3 and of Sc-stabilized cubic ZrAl3 precipitates [J]. Phys Rev B, 1990, 41: 12553-12561.
[43] Xu J H, Oguchi T, Freeman A J. Crystal structure, phase stability, and magnetism in Ni3V [J]. Phys Rev B, 1987, 35: 6940-6943.
[44] Gou H Y, Hou L, Zhang J W, Li H, Sun G F, Gao F M. First-principles study of low compressibility osmium borides [J]. Appl Phys Lett, 2006, 88(22): 221904.
[45] Chiodo S, Gotsis H J, Russo N, Sicilia E. OsB2 and RuB2, ultra-incompressible, hard materials: First-principles electronic structure calculations [J]. Chem Phys Lett, 2006, 425(4-6): 311-314.
������Ľṹ����ѧ�͵������ʵĵ�һ��ԭ���о�
�ƺ���1��κ Ⱥ2������÷3���� ƽ4
1. ��������ѧԺ ��ѧ����ϵ������ 721013��
2. �������ӿƼ���ѧ ����ϵ������ 710071��
3. ��������ѧԺ ����ϵ������ 721007��
4. ������ѧ ����ϵ������ 710069
ժ Ҫ�����û����ܶȷ������۵ĵ�һ��ԭ�����㷽���о��˹��������������(Ti2B, TiB��TiB2)�ľ����������ѧ���ܺ͵������ʡ���������ļ�������ʵ��ֵ�Լ��������ۼ���ֵ�Ǻϵúܺá����Գ����ļ�������������3�������ڳ�ѹ�¶�����ѧ�ȶ��ġ�����������(120 GPa)��3��������嵯ģ�����ܴ����������������Ӷ����ӣ���������ɽ����������������ԭ������ԭ��֮���γɶ��ۼ�����ġ���TiB2�⣬TiB���нϴ���嵯ģ���ͼ���ģ������������Ϊһ�־������ÿ�ѹ���ʵ�Ӳ�ʲ��ϡ����⣬���⼸������ĵ��Ը����������ʺ͵°��¶�Ҳ���������ۡ�����̬�ܶȺ�ԭ�ӵ�ɲ��ӷ�����������3������Ļ�ѧ���ɷ�ͬʱ�����˹��ۡ����Ӻͽ����ɷ֡�
�ؼ��ʣ��������һ��ԭ�����㣻��ѧ���ʣ�̬�ܶȣ�Mullikenԭ�Ӳ��ӷ���
(Edited by YANG Hua)
Foundation item: Project (2010JK404) supported by the Education Committee Natural Science Foundation of Shaanxi Province, China; Projects (ZK0918, ZK0915) supported by the Baoji University of Arts and Sciences Key Research, China
Corresponding author: YAN Hai-yan; Tel: +86-917-3566589; E-mail: hyyan1102@163.com; WEI Qun; Tel: +86-917-3364258; E-mail: weiaqun@tom.com
DOI: 10.1016/S1003-6326(11)60906-0
[1] LUNDSTORM T, RUNDQVIST S. Borides, silicides and phosphides [M]. London: Methuen, 1965.
[9] Post B, WGlaser F, Moskowitz D. Transition metal diborides [J]. Acta Metall, 1954, 2(1): 20-25.
[10] Gilman J J, Roberts B W. Elastic constants of TiC and TiB2 [J]. J Appl Phys, 1961, 32(7): 1405.
[23] Bl?uml P E. Projector augmented-wave method [J]. Phys Rev B, 1994, 50(24): 17953-17979.
[28] Wyckoff W G. Crystal structures [M]. New York: Wiley, 1963.
