
���±�ţ�1004-0609(2009)10-1759-07
���������ɱ�����ȶ��Լ���
�������������ţ��ƽ�����������
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ ��ɫ�������Ͽ�ѧ�빤�̽������ص�ʵ���ң���ɳ 410083)
ժ Ҫ��
���õ�һԭ������ƽ�沨����������������ѧ���ɱ���(100)��(110)��(111)����������������ӽṹ�����ݱ����ܼ���Ԥ�������ɱ���ṹ���ȶ��ԡ���������������ɱ���ṹ�ȶ�����ǿ������˳��Ϊ(111)��(100)��(110)������ԭ�ӳ�ԥ����������漸�νṹ�ı仯������ʹ�����ĵ��ӽṹ��������Է����ı䣻(100)��(110)��(111)�����ԥ�ֱ�Ϊ3.337%��-6.147%��-2.364%���������ܶȲ�ͬ��������ܲ��죬����ԭ�Ӳ�ʹα���ԭ�Ӳ�ĵ�����ܶ���s��p��������·ֲ�������ԭ�Ӳ����ܶ�Խ������Խ�͡�
�ؼ��ʣ�
Al��������һԭ���������ԥ����������
��ͼ����ţ�TG146.1 ���ױ�ʶ�룺A
Calculation of stability of free surfaces in aluminum crystal
ZHANG Xin-ming, LIU Jian-cai, TANG Jian-guo, CHEN Ming-an
(Key Laboratory of Non-ferrous Metal Materials Science and Engineering, Ministry of Education, School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: The surface energy, atomic geometry and electronic structures of Al(100), (110) and (111) free surfaces were calculated using the method of supercell and the first-principles pseudopotential plane waves within generalized gradient approximation. According to the calculated surface energy, the structural stability of Al free surfaces from strong to weak is predicted in the order as (111), (100) and (110). The relaxation of the surface atom layers not only causes the change of geometrical structures of the surface models, but also leads to the variation of their electronic structures and bonding characters. For the (100), (110) and (111) free surfaces, the calculated surfaces relaxation are 3.337%, -6.147% and -2.364%, respectively. The surface energy is related to the surface electron density distribution, the electron density of orbital s and p of the first two surface atom layers redistributes. The higher the surface electron density is, the lower the surface energy is.
Key words: aluminum crystal; first principle; surface relaxation; surface energy
�����������ǵ�����������ҪԪ��֮һ���ڵ�����Ϣ�豸������������ͼҵ�Ȳ�Ʒ�����Ų����������Ҫ���á����ŵ���������С�ͻ���չ����ȻҪ������������������������С[1]������ͨ��̽�����о����֣������绯ѧ�ķ�����������{100}�����港ʴ�������������ף����Դ������������ı�����ͱȵ���[2]����ˣ�����һֱ��ȡ���ִ�ʩ���Ӹߴ�����������֯��{100}<001>��ռ���ʣ�ʹ֮����95%[3-4]��������Ȼ��������߸�ѹ����{100}��ռ���ʣ����������в�������{100}ƽ��������Ľṹ�����Ҵ����������͵ľ���ѧƽ��ṹ����{110}��{111}�ȡ���ͬ�ľ���ѧƽ����ڸ�ʴ���̾��в�ͬӰ��[5]����ˣ�����������ͬ����ѧƽ��Ļ��Ե��о��Ե÷dz���Ҫ��
��Ȼ�����Ѿ���չ�˺ܶ�ⶨ����ԭ�ӽṹ��ʵ�鷽��[6]��ëϸ�ܷ����ٽ�������������㷨�ȣ�������ֵ����Һ��Ƚ�ȷ���Թ������кܴ�ľ����ԣ�������������������������������IJ�һ�¶�����һ�����졣ͬʱ�����ھ��������и������ԣ�ʵ�鷽�������������ĸ��������ı����ܡ�����ͨ�����۵ķ�������ñ����������
���������ܶ�ѧ�߲��ò�ͬ�ļ��㷽���Բ�ͬ�ľ������ṹ�����˼���[7-9]��������Al���������ȱ��ϵͳ��ȫ����о�[10-13]������Щ����������ڱ����������������ǵĹ�ϵ�������ٿ��DZ����ԥ�Խṹ��Ӱ�졣�������߲��õ�һԭ���ļ��㷽�����������г�����3������ѧƽ��{100}��{110}��{111}�Ĺ��͡���ԥ��������ӽṹ�������ܽ����˱Ƚ�ϵͳ���о����ý�����˽������������ʼ���һ����ʾ�������治ͬ�����ڸ�ʴ�����е������ṩ�˲ο���
1 ��ͬ����ѧ����ṹ�ȶ��Ե�����ѧ����
�����ܿ�������Ϊ���������������¾������������������������Сȡ���ڱ���ԭ�Ӽ������ã������ԭ�ӵļ��νṹ������ء��������ӵľ��壬�ɲ�������ѧ-��ѧ����[14]�������ж������ṹ�ȶ��ԡ�
��һ���������NA��ԭ�ӣ�ÿ��ԭ����Z����λԭ�ӣ�����ZNA/2����ϼ�������ÿ����ϼ��ļ���Ϊ��AA�������������?HS�����ƶ����м�������������
![]()
����ij���ض����棬������ԭ��������ԭ���������n����ϼ�������n���������ڱ��棬���������?U(��������)�ɹ������£�
![]()
ʽ�У�NSΪ��λ�����ϵ�ԭ������FCC��������ÿ��ԭ�ӵ������ԭ����ΪN=12�����γ�(111)���棬�����ԭ��ɥʧn=3����ϼ���������3�������Ҽ����� (111)����Ӧ�ı�����?U(111)�͵�λ�����ϵ�ԭ���� ���£�
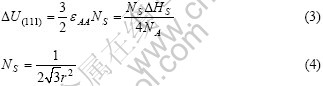
ͬ���ɵã�����(110)�棺

����(110)�棺

��ˣ���3������������ı����ܱ�ֵ���£�

��������ԵĽ����֪����Ȼ������ı���ʽ�в���ֱ�Ӽ����FCC����ƽ��ı����ܣ����ɴ�ȴ���Զ������ֵ�Ĵ�С���бȽϡ������ʾ��������ı������ɴ�С˳��Ϊ(100)��(110)��(111)��(100)��ı��������ߣ�(111)��ı����ȶ�����á�
����ѧ������Ϊ��������������������������Ϊ����棬�Դﵽ���ȶ��Ľṹ����Ϊ�������Žṹ��(111)���������棬(111)���ԭ�����ܶ���14.1 nm-2��(100)���ԭ�����ܶ���12.3 nm-2��(110)���ԭ�����ܶ���8.7 nm-2����ԭ�����ܶȿ�����Ϊ��������������ɴ�С˳��Ϊ��(110)��(100)��(111)����ǰ���û�ѧ���㷨������������Ϊ��ѧ����ֻ��һ�����ԵĽ��ۡ����ڲ�ͬ�����ԭ�ӣ�������ԭ�ӽṹ������������ӽṹ���ڲ��죬����ԭ�ӵļ��κ͵��ӽṹ�ᷢ���仯������ı�����ֵ��������ͬ�����⣬������ļ����У�������ܵļ���������Ƶؼ������䡰��ѧ������ã���û�п��DZ����ԥ���ع��ȡ��ṹ���Ӱ�졣Ϊ�˽�һ����ʾAl���岻ͬ����Ľṹ�ȶ�������漸�κ͵��ӽṹ�Ĺ�ϵ���������߶�Al����ı���ṹ���������������
2 ����ģ���뷽��
������û����ܶȷ������۵�����ƽ�沨����-CASTEP(Cambridge serial total energy package)���������[15]�������ݶȽ���(GGA)�Ľ�������(PBE)�ij�������(Ultrasoft)����������ʵ��۵���֮��������(3s23p1)���ṹģ�Ͳ���BFGS�㷨�����Ż����ԥ�������в�����ά�����Ա߽������ij�����ģ��ģ����棬Ϊ�˱�֤z����ԭ�Ӳ㲻����ã�����Ҫ���㹻�����ղ㣬һ����Ϊ����պ��Ϊ10?����������Ҫ�������ھ����У�ԭ�Ӳ����Ķ����Ǿ���������Ч�Ե�һ����Ҫ���أ�ͨ����Ϊ5��ԭ�Ӳ����㹻����Ч�������Ҫ���Ҫ��ԭ�Ӳ���֮�⣬��Ҫ���к��ʵij�ʼ�̶��������Ա�֤�����ԭ���ܹ���ֳ�ԥ��ͨ��ԭ�Ӳ�������ղ��Ȳ��ԣ���(100)��(110)��(111)����ģ�͵�ԭ�Ӳ㶼ȡ7�㡣����ʱ�̶���������ײ��3��ԭ�ӣ�����ģ�ͼ�ͼ1��ʾ���ڼ���ʱ���ܽضϵ�ȡ350 eV������Ԩ�����ֲ���Monkhorst-Pack��ʽ������K�㷽����FFT����ȡ (24��24��24)����ӦAl(111)�� (110)��(100)����ģ�ͣ�K��ֱ�ȡ(10��10��1)��(6��9��1)��(9��9��1)����Ǣ����ʱ�����е�����������ֵ��Ϊ1.0��10-5 eV/atom��ÿ��ԭ���ϵ���С��0.03 eV/?������ƫ��С��0.001 ?��Ӧ��ƫ��С��0.05 GPa��
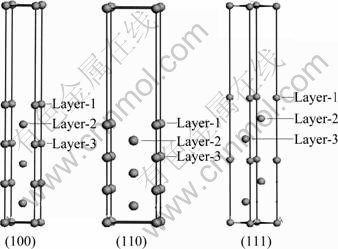
ͼ1 ��(100)��(110)��(111)�ı���ģ��
Fig.1 Surface models of (100), (110) and (111) of aluminum crystal
3 ����������
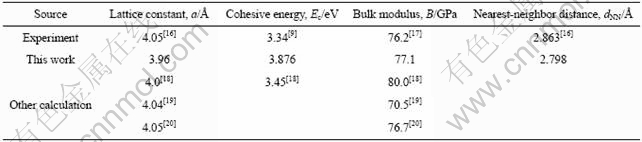
Ϊ����֤�������Ŀɿ��ԣ���������������4����ԭ����ɵ�FCC-Al����ľ�����a�������Ec����ģ��B�������ԭ�Ӽ��dNN������ʵ��ֵ�������ļ����������˱Ƚϣ�������1���С����������Al�ľ�����a��Ec��B��dNN��ʵ��ֵ�����ϽϺã�˵������ģ�Ϳ��С�
��1 Al�ľ�����a�������Ec����ģ��B�������ԭ�Ӽ��dNN
Table 1 Lattice constant a, cohesive energy Ec, bulk modulus B and nearest-neighbor distance dNN of atoms for Al crystal
�ñ���ṹ���г�ֳ�ԥ�����������ڱ����ԭ���ϵ�Ӧ�����Ӷ��õ������ȶ��Ľṹ������������ı���ԭ���ڳ�ֳ�ԥ�����£������ϲ������ع������Ա���������Ҫ�ǿ������ԭ�ӵij�ԥ����Ϊ�˶�������Al����ԭ�ӳ�ԥ������������£�

��2����ΪAl����ģ��ԭ�ӳ�ԥ�ļ��νṹ�����仯������d12Ϊ��������2��֮���࣬d23Ϊ��2�����3��ļ�࣬d34Ϊ��3�����4��ļ�ࡣ
��2 Al����ģ�͵ļ��νṹ����
Table 2 Geometric structural parameters of Al surface models
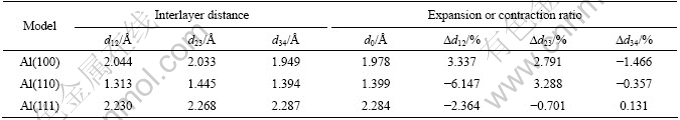
�ɴ˿��Կ�����3����ͬ�����������ڲ�֮����ڲ�ͬ�̶ȵij�ԥ�������FCC-Al������ԣ�(100)�������������������չ����(110)��(111)���㶼������������Al(111)ģ�͵ĵ�2�����3��ԭ�Ӽ�IJ�����Ȼ���٣���������������Al(110)��Al(100)ģ�͵ĵ�2�����3��ԭ�Ӽ�IJ��������������͡����⣬�ӱ�2�����Կ���|?d12|��|?d23|>|?d34|��������ԭ�ӳ�ԥЧӦ�ӱ����1�㵽��4��������Խ���ڸ����ڲ�֮���仯ԽС��Al(110)����ԭ�ӵij�ԥ�̶Ȳ����Al(111)���Al(100)���2������Al(100)�����ֱ�Al(111)����ԭ�����������̶ȴ���Al(110)����ԭ�ӵij�ԥ�̶����Al(100)�����֮��Al(111)������С����˵��Al(111)��������ǽ��ȶ��ı��档
��������һ����Ҫ����������ͨ�����������ܣ����Խ�һ���˽�����ȶ��Ժͱ�������������������Ϣ������ֻ���ǻ�̬(0 K)����������Ա����ܵļ���ʽ�������£�

���ڱ�����������ǻ�̬ԭ������(p=0 Pa��T=0 K)�����pV��TS���Ժ��Բ��ƣ����������3���С��ӱ�3��֪��Al(111)�����ܵļ�������ʵ��ֵ�Ե�һЩ������Ҫ�����ڱ�����ı�����û�п����¶ȶԱ����Ӱ�졣����õ��ĸ�������ı����ܵĴ�С˳��ͷ��Ӷ���ѧ������һ�£�����Al(110)���������Al(100)�����ܴ�֮��Al(111)��������ͣ����Al(111)�������ȶ���������Al(100)������У�Al(110)������ȶ���������ߡ�ֵ��ָ�����ǣ��ӵ�һԭ���������õ�Al(111)��(100)��(110)�ı����ܱ仯����(��EAl(111)?EAl(100)?EAl(110)= 1?1.274?1.321)���������ѧ-��ѧ��������(��EAl(111)?EAl(100)?EAl(110)=1?1.115?1.021)����ȣ���Ȼ��ֵ�����仯����ȴ����ȫ��ͬ��������ѧ-��ѧ����Ԥ��Al(100)����Ļ�����ߣ����ӵ�һԭ���ļ���ȴ�������������õ���Al (110)���档��һԭ��������˳�������ԭ���ܶ�˳��һ�£�����ԭ���ܶ�Խ�ߣ�������Խ�ͣ���һԭ�����������ӷ���ʵ�������
��3 Al����ı������
Table 3 Surface parameters of aluminum crystal
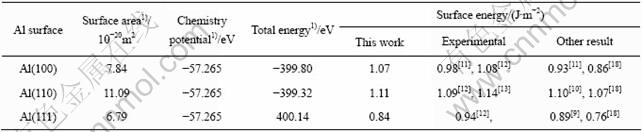 1) Results of this work.
1) Results of this work.
Ϊ�˽�һ��˵�������岻ͬ����Ľṹ�ȶ��Բ���ĵ��ӻ��ƣ��������˱���ģ�͵ĵ�����̬�ܶȺͱ����һ����������ľ���̬�ܶ�(LDOS)����ֲ�̬�ܶ�(PDOS)(��ͼ2��ʾ)����ͼ2��֪��Al�����м۵���̬�ܶ���Ҫ��3s��3p�ֲ�̬�ܶȹ��ɣ����о�����p���ӹ��ף������ܼ�(EF)�����������Ե�����϶���Ƚ�Al 3���������������ܶȷֲ���֪����Al(111)�����������γɱ���ģ�͵���̬�ܶ��������Al����ı䲻����Al��������϶����Լ4 eV������ģ��ȴ�³�����һС�ı���̬��������Al(100)���������ԣ���Al(100)��Al(110)������һ�������˷����ܼ����ļ۵��ӷ壬ʹ����϶���۵����ܶ�����̧�ߡ���Ȼ��������Щ�ڸ��ܼ��������ı���̬����ܼ������ٵĵ���̬�ܶ�ʹ����ṹ���ڱȾ���ṹ������Խϸߵ�״̬����������˱����ܣ�������������Է���������˱��������ȶ�������ԡ�ʵ���ϣ��������Ҳ�ɴӼ���ģ�ͷ����ܼ�����ֵ��С�ıȽ��еõ�����˵����Al�����EF=8.504 eV��Al(111)�� Al(110)��Al(100)�����EF�ֱ�Ϊ3.005��1.306��2.401 eV������ھ��壬����ķ����ܼ��ͣ���˱�����ȶ��ԱȾ���ͣ�����ͬ�����У��ȶ�����õ���Al(111)�棬Al(100)���֮���ȶ�����������Al(110)�档
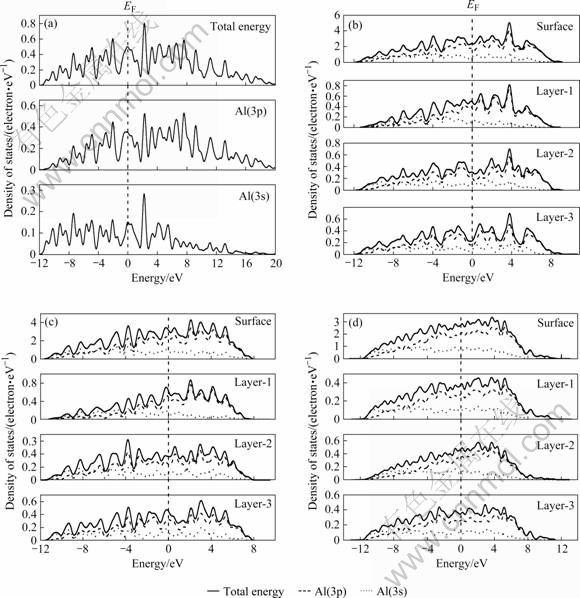
ͼ2 Al���������ĵ���̬�ܶ�ͼ
Fig.2 Density of states of aluminum crystal and surfaces: (a) Al bulk; (b) Al(100); (c) Al(110); (d) Al(111)
��һ����Al���治ͬԭ�Ӳ�ľ������̬�ܶȽ��бȽ�ʱ���֣������ԭ��(Layer-1)�ľ������̬�ܶ���������ڲ�ԭ��(Layer-3)�����˺ܴ�仯������ԭ�Ӳ��ϵ�����ܶ�Al(111)����ͣ�Al(110)����ߡ���Ȼ��������Щ��������̬�ܶȵı仯�����˲�ͬ����ṹ�ȶ��ԵIJ��졣��һ���Ƚϲ�ͬԭ�Ӳ�������̬�ܶȲ��ѿ������Ա�����ӽṹ���Ӱ�����Ҫ��Layer-1��Layer-2���Ƚ�Layer-2��Layer-3�ľ������̬�ܶȿɼ���Layer-2���ԭ��Ҳ���ڽϸߵĵ�����̬��������нϸߵĻ��������ˣ���Ҳ������ѧ-��ѧ��������ȷԤ�ⲻͬ����ṹ�ȶ��Ե�һ��ԭ������ԭ�Ӳ�Զ���������������룬ԭ�Ӳ��LDOS����̬����Խ��Խ���ԣ�ͬʱ����̬Ѹ��˥����
LDOS�����ʾ�ھ���̬�ܶ��У�3p̬�ܶ�ռ����Ҫ�ijɷ֣����ڸ�ԭ�Ӳ�ľ���̬�ܶ��У�3s���Ӿ������ڼ۴��ĵ��ܶˣ��Է����ܼ�������̬�ܶȼ������ף���ˣ�����̬����ȫ����3p̬�����ס���һ���棬��3s̬�ܶȶ��ԣ��ڶ�ԭ�Ӳ��е�3s�ֲ�̬�ܶȾ���������̬�е�3s�ֲ�̬�ܶȣ����֮�£������3p�ֲ�̬�ܶ�����̬��3p�ֲ�̬�ܶȵ�����ȴ���ö࣬����ζ��Al�е�3p���Ӷ�Al�����ijЩ�������������á���ϸ�رȽ�ͼ�����Ӧ�ľ���̬�ܶȼ���ֲ�̬�ܶȿ��Է��֣�3s̬�ܶȵķֲ�������ͬ������̬�ܶȵIJ�����Ҫ������3p̬����˵����Al����ṹ�ij�ԥ��Ҫ��3p�����йء�
4 ����
1) Al����������ɴ�С˳��Ϊ(110)��(100)��(111)��(111)��Ľṹ�ȶ�����ߣ�(100)���֮��(110)����ȶ���������ͬ�����£�Al(110)���������ߣ��������������ʷ�Ӧ��(111)����������ȶ���
2) Al(110)�������������̶�ԼΪ��Al(111)����Al(100)���2��������ԭ�ӳ�ԥ����������漸�νṹ�ı仯����ʹ�����ĵ��ӽṹ��������Է����ı䣬ʹ�����ԭ�ӵĵ���̬�ܶȷ��ͷ����˱仯��������ֵ�ɱ���������ԭ�Ӳ�ĵ�����ܶ���s��p����ϵķֲ����������������ܶ�Խ�ߣ�������Խ�͡�
REFERENCES
[1] MAO W M, JIANG H, YANG P, FENG H P, YU Y N. Distribution of microelements and their influence on the corrosion behavior of aluminum foil[J]. Journal of Materials Science & Technology, 2005, 21(1): 43-46.
[2] XIAO Y Q, LIU C M, JIANG S, CHEN Z Y, ZHANG X M. Influence of high hot-rolling temperature and multistage annealing upon the cube texture of high purity aluminum capacitor foils[J]. Textures of Materials, 2002, 408/412(4): 1449-1452.
[3] ZHANG X M, LIU S D, TANG J G, ZHOU Z P. Mechanism of strengthening of cube texture for high purity aluminum foils by additional-annealing[J]. Trans Nonferrous Met Soc China, 2003, 13(3): 499-503.
[4] LIU C M, ZHANG X M, CHEN Z Y, DENG Y L, ZHOU Z P. Evolution of recrystallization textures in high voltage aluminum capacitor foils[J]. Trans Nonferrous Met Soc China, 2001, 11(4): 513-516.
[5] �� ��. �Ǹ���ϵ���������ø�ѹ�����������о�[D]. ����: �����Ƽ���ѧ, 2007.
YANG Hong. Research on the non-chromic acid high voltage anode aluminum foil for electrolytic capacitor[D]. Beijing: University of Science and Technology Beijing, 2007.
[6] RTYSON W, MW A. Surface free energies of solid metals: Estimation from liquid surface tension measurements[J]. Surf Sci, 1977, 62(1): 267-276.
[7] METHFESSEL M, HENNING D, SCHEZER M. Trends of the surface relaxations, surface energies, and work functions of the 4d transition metals[J]. Phys Rev B, 1992, 46(8): 4816-4829.
[8] KOLLAR J, VITOS L, SKRIVER H L. Surface energy and work function of the light actinides[J]. Phys Rev B, 1994, 49(16): 11288-11292.
[9] RODRIGUEZ A M, BOZZOLO G, FERRANTE J. Multilayer relaxation and surface energies of fcc and bcc metals using equivalent crystal theory[J]. Surf Sci, 1993, 289(1/2): 100-126.
[10] WANG X C, YU J, QIAN K Y, WANG F, MA J X, HU X. The calculation of the surface energy of high-index surfaces in metals at zero temperature[J]. Surface Science, 2004, 551(3): 179-188.
[11] MUTASA B, FARKAS D. Atomistic structure of high-index surfaces in metals and alloys[J]. Surface Science, 1998, 415(3): 312-319.
[12] SCHOCHLIN J, BOHNEN K P, HO K M. Structure and dynamical at the Al(111)-surface[J]. Surf Sci, 1995, 324(2/3): 113-121.
[13] RAEKER T J, DE PRISTO A E. Corrected effective-medium method. IV. Bulk cohesive and surface energies of second-and third-row metals and multilayer relaxation of Al, Fe, and Ni[J]. Phys Rev B, 1989, 39(14): 9967-9982.
[14] Ф����, �����. ��������ѧ[M]. �Ϻ�: �Ϻ���ѧ����������, 1999: 417.
XIAO Ji-mei, ZHU Feng-wu. Material energetics[M]. Shanghai: Shanghai Science and Technology Press, 1999: 417.
[15] SEGALL M D, LINDAN P, PROBERT M J. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Condense Matter, 2002, 14(11): 2717-2744.
[16] STRAUMANIS M E, WOODARD C L. Lattice parameters and thermal expansion coefficients of Al, Ag and Mo at low temperatures[J]. Acta Cryst A, 1971, 27: 549-551.
[17] TALLON J L, WOLFENDEN A. Temperature dependence of the elastic constants of aluminum[J]. J Phys Chem Solids, 1979, 40(11): 831-837.
[18] �ŷ�Ӣ, ��ӢԪ, ����ϼ, ��ʥ��. Al(001)��Al(110)��Al(111)������ܵ��ܶȷ������ۼ���[J]. ��ʴ��ѧ���������, 2005, 17(1): 47-49.
ZHANG Fang-ying, TENG Ying-yuan, ZHANG Mei-xia, ZHU Sheng-long. Density functional theory study of surface energies of Al(001), (110) and (111)[J]. Corrosion Science and Protection Technology, 2005, 17(1): 47-49.
[19] KIEJNA A, LUNDQVIST B I. First-principles study of surface and subsurface O structures at Al(111)[J]. Physical Review B, 2001, 63(8): 085405(1-10).
[20] ZHUKOVSKII Y F, JACOBS P W M, CAUS? M. On the mechanism of the interaction between oxygen and close -packed single- crystal aluminum surfaces[J]. J Phys Chem Solids, 2003, 64(8): 1317-1331.
������Ŀ�������ص�����о���չ�ƻ�������Ŀ(2005CB623706)
�ո����ڣ�2008-10-27�������ڣ�2009-01-03
ͨ�����ߣ������������ڣ���ʿ���绰��0731-8830265��E-mial: jcliu1568@yahoo.com.cn
