
Trans. Nonferrous Met. Soc. China 31(2021) 853-864
Correlation between mixing enthalpy and structural order in liquid Mg-Si system
Jin WANG1, Jing-yu QIN2, Ji-xue ZHOU1, Kai-ming CHENG1, Cheng-wei ZHAN1, Su-qing ZHANG1, Guo-chen ZHAO1, Xin-xin LI3, Ke-chang SHEN4, Yi ZHOU5
1. Shandong Provincial Key Laboratory of High Strength Lightweight Metallic Materials, Advanced Materials Institute, Qilu University of Technology (Shandong Academy of Sciences), Ji��nan 250014, China;
2. Key Laboratory for Liquid-Solid Structural Evolution and Processing of Materials, Ministry of Education, Shandong University, Ji��nan 250061, China;
3. School of Materials Science and Engineering, Liaocheng University, Liaocheng 252000, China;
4. Ulsan Ship and Ocean College, Ludong University, Yantai 264025, China;
5. School of Mathematics and Physics, Qingdao University of Science and Technology, Qingdao 266061, China
Received 3 April 2020; accepted 28 November 2020
Abstract:
The mixing enthalpies and structural order in liquid Mg-Si system were investigated via ab-initio molecular dynamics at 1773 K. By calculating the transferred charges and electron density differences, the dominance of Si-Si interactions in the chemical environments around Si was demonstrated, which determined that the mixing enthalpy reached the minimum on Mg-rich side. In terms of Honeycutt and Anderson (HA) bond pairs based on the partial pair correlation functions, the attraction between Si-Si pairs and Mg atoms was revealed, and the evolution of structural order with Si content was characterized as a process of constituting frame structures by Si-Si pairs that dispersed Mg atoms. Focusing on tetrahedral order of local Si-configurations, a correlation between the mixing enthalpy and structural order was uncovered ultimately, which provided a new perspective combining the energetics with geometry to understand the liquid Mg-Si binary system.
Key words:
liquid Mg-Si system; mixing enthalpy; structural order; ab-initio molecular dynamics;
1 Introduction
As a kind of traditional lightweight high strength material, Mg-Si alloys have regained attention owing to their interesting mechanical and thermal properties [1]. The mechanical properties of Mg-Si alloys are similar to those of natural bone, what is more, the excellent biodegradability and biocompatibility make them promising candidates for orthopedic clinical applications [2]. Besides, Mg-Si alloys have considerable high thermal conductivities and low coefficients of thermal expansion, which are essential properties for applications in the electric industry [3]. Therefore, in order to promote the upgrading of implant and electronic products, it is necessary to design the capable Mg-Si alloys based on a comprehensive understanding on this binary system.
Mg-Si binary alloy phase diagram provides an important guideline for the alloy design. Since the first experimental determination performed by VOGEL in 1909 [4], Mg-Si binary alloy phase diagram has been optimized more than 7 times [5-11]. One of the major difficulties is to obtain the accurate thermodynamic description on the liquid phase, because no direct measurement for the mixing enthalpy has been available in liquid Mg-Si system so far. In 1967, ELDRIDGE et al [12] firstly derived the mixing enthalpies of liquid Mg-Si alloys from the measured activities by an isopiestic method. However, the isopiestic experiments were limited to the composition below 60 at.% Si. Combining the isopiestic experiments of ELDRIDGE et al [12] with the phase diagram information, GEFFICEN and MILLER [13] reevaluated the mixing enthalpies later, which show visible differences from the results of ELDRIDGE et al [12].
In an effort to eliminate this confusion, YUAN et al [11] described the excess Gibbs energies of the liquid phase by using Kaptay equation [14,15], and ELDRIDGE et al��s data were reproduced. On the other hand, JUNG et al [10] critically evaluated all the available thermodynamic and phase diagram data in Mg-Si binary system. With the aid of the extended modified quasi-chemical model [16,17] for short-range order in the liquid phase, their optimized mixing enthalpies are close to the data given by GEFFICEN and MILLER [13]. Although the confusion continues, JUNG et al��s work [10] suggests that the mixing enthalpies in liquid Mg-Si system are worth further understanding combined with the structural order.
Unfortunately, investigations on the liquid structures of Mg-Si alloys are not sufficient. Recently, LIU et al [18] have performed ab-initio molecular dynamic (AIMD) simulations to investigate the structures of a series of liquid Mg-Si alloys at the temperatures 100 K higher than the liquidus. By examining chemical short-range order around Si atoms, a maximum was identified at eutectic Mg47Si53 alloy. Further, QIN et al [19] emphatically studied atomic self-diffusion behaviors of liquid Si-contained metallic systems including Mg-Si, and two main factors on the self-diffusion coefficient of Si (DSi) were confirmed. The dominant factor is the partial coordination number ZSi-Si: the larger the ZSi-Si is, the smaller the DSi will be; the secondary one is medium-range order: the stronger the medium- range order is, the smaller the DSi will be. On this basis, the coupling diffusion behavior between Mg and Si atoms in liquid Mg-Si system below 60 at.% Si was identified.
These reported AIMD works provide insights into the structural order in liquid Mg-Si system. However, two independent variables affecting atomic behaviors are involved: one is composition and the other is the temperature. For the sake of understanding the mixing enthalpy combined with structural order, composition should be the only independent variable, which is difficult to be extracted accurately from the above works by eliminating the temperature effect. In this work, the mixing enthalpies and structural order in liquid Mg-Si system were investigated at a constant temperature via AIMD. The transferred charges and electron density differences were calculated to explore the atomic interaction mechanism in the evolution of the mixing enthalpy, and the structural order was characterized by analyzing Honeycutt and Anderson bond pairs based on the partial pair correlation functions. Finally, a correlation between the mixing enthalpy and structural order was uncovered in liquid Mg-Si system focusing on tetrahedral order of local Si-configurations.
2 Methodology
2.1 AIMD simulation
The AIMD simulations were performed by the Vienna ab-initio simulation package (VASP) [20]. The projector augmented wave potentials [21] were adopted to determine the interaction between ion cores and valence electrons, and the electronic exchange-correlation was described by the generalized gradient approximation (GGA) [22] with PW91 functionals [23]. Only ��-point was used for sampling the Brillouin zone. NVT ensemble was employed by means of Nos��-Hoover thermostat [24,25] to control the temperature, and Newton��s equations of motion were solved via the velocity Verlet algorithm [26] with a time step of 3 fs.
Because the melting point of Si (1687 K) is even higher than the boiling point of Mg (1380 K), it is difficult to determine a constant simulation temperature for ensuring that pure Mg and Si are both in the liquid state. Considering that the random atomic distribution is a common character shared by gases and liquids, we firstly established an initial configuration of liquid pure Mg via Monte-Carlo algorithm, which contains 240 Mg atoms distributed randomly. The initial configuration was relaxed for 9 ps to reach the equilibrium state under an average external pressure of (0��50) MPa at 973 (50 K above the melting point of Mg), 1073, 1173, 1273, and 1773 K (393 K above the boiling point of Mg and 86 K above the melting point of Si), respectively. The calculated molar volumes are exhibited in Fig. 1. It can be seen that the molar volume linearly evolves with the temperature, and there is no abruption corresponding to the first order phase transformation in this temperature region. Hence, we set 1773 K as the simulation temperature in this work, and the simulated liquid Mg-rich alloys can be considered as overheating melts.

Fig. 1 Molar volume of liquid pure Mg at (0��50) MPa
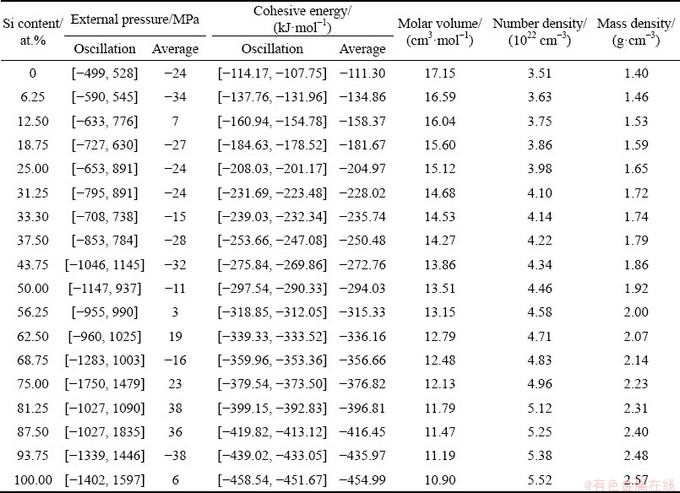
After determining the simulation temperature, 18 composition points were sampled from pure Mg to Si. The initial configuration for each composition point, which contains 240 atoms, was also established by Monte-Carlo algorithm and relaxed for 9 ps to reach the equilibrium state under an average external pressure close to 0 at 1773 K. Following the relaxed configurations, AIMD runs were continued for 9 ps to obtain the average physical parameters and structural order of liquid Mg-Si alloys. The obtained physical parameters, including external pressures, cohesive energies, number densities, and mass densities, are given in Table 1. Due to the atom vibrations, the external pressures and cohesive energies are always oscillated. The oscillation ranges are provided in Table 1, and each of the average values is derived based on 3000 configurations in the continued 9 ps run. Among these physical parameters, only the mass density of liquid pure Si can be verified because of the lack of experimental data. The calculated result of 2.57 g/cm3 at 1773 K is a bit smaller than the experimental value of 2.59 g/cm3 at 1733 K [27], revealing the accuracy of the current AIMD simulations.
Table 1 Physical parameters of liquid Mg-Si alloys at 1773 K
2.2 Data analysis
The mixing enthalpy (��H) is defined as Eq. (1):
��H=Halloy-(1-cB)HA-cBHB (1)
where cB is the concentration of component B in the liquid binary alloy, and Halloy, HA, and HB denote the enthalpies of the alloy, pure A, and pure B, respectively. Because each sample in this work was relaxed under an average external pressure close to 0 at 1773 K, the enthalpy is approximate to the cohesive energy as listed in Table 1.
The electron density differences are calculated from Eq. (2):
����=��actual-��sphericalized (2)
where ��actual denotes actual electron density in an alloy, and ��sphericalized denotes the superposed sphericalized electron density in the configuration same with the considered alloy. With the aid of Bader Charge Analysis [28], the total charge of a single atom in an alloy is found by integrating the actual electron densities within the ��Bader�� region, and the transferred charge can be obtained by subtracting the total charge in the free state from that in the alloy.
The partial pair correlation function (PPCF, gA-B(r)) is derived from Eq. (3) [29]:
 (3)
(3)
where V is the total volume, NA and NB are the atomic numbers of A and B, respectively, niB(r,��r) is the atom number of B in the sphere shell around the ith A atom from radius r to radius r+��r. Further, the partial coordination number is provided by the following Eq. (4):
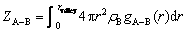 (4)
(4)
where rvalley denotes the position of the first valley in gA-B(r), and ��B is the number density of B.
Honeycutt and Anderson (HA) bond pair [30] characterizes the topological short-range order by a sequence of three integers as ��ijk��: the first integer i is labeled as 1 if the atoms comprising a root pair are near-neighbors; the second integer j is the number of near-neighbors shared by the root pair; the third integer k is the number of bonds among the shared neighbors. It should be noted that, the ��pair�� and ��bond�� are identified by the fact that the atoms are within a cutoff distance corresponding to the position of the first valley in the PPCF, instead of the atomic interaction.
The composition of shared neighbors reflects the atomic distribution behavior around a root pair. To describe these behaviors, we defined a parameter 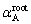 by referring to Ref. [31]:
by referring to Ref. [31]:
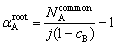 (5)
(5)
where 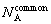 denotes the atomic number of A in shared neighbors. The parameter states three distribution behaviors of A: is equal to zero for the ��random�� distribution; the positive means the attraction between the root pair and A; the negative one means the repulsion.
denotes the atomic number of A in shared neighbors. The parameter states three distribution behaviors of A: is equal to zero for the ��random�� distribution; the positive means the attraction between the root pair and A; the negative one means the repulsion.
The number of bonds among the shared neighbors reflects the dense degree of the atomic arrangement around a root pair. Herein, we defined a parameter ��root to quantify the dense degree as follows:
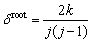 (6)
(6)
where k is number of bonds among the shared neighbors. It should be clarified that the denominator j(j-1)/2 means the ideal upper limit of the number of bonds among the shared neighbors, and the parameter ��root is only applicable for comparing the dense degrees between the bond pairs with the same j. For example, both 120 and 121 bond pairs can be searched out in liquid pure Mg and the maximum ��root is 1 for j=2, while ��root for j=5 allows a maximum of 0.8 corresponding to the bond pair 158.
Finally, a local Si-configuration in this work is composed of five Si atoms and formed by a central one with its 1st, 2nd, 3th, and 4th near-neighbors. Its tetrahedral order is evaluated by the parameter qt [32]:
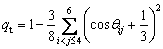 (7)
(7)
where ��ij is the angle formed by an atom with its ith and jth near-neighbors, and qt=1 means perfect tetrahedral order.
3 Results and discussion
3.1 Mixing enthalpy
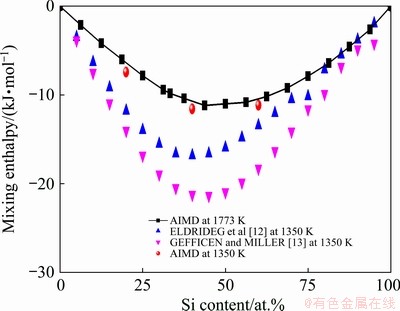
Fig. 2 Calculated mixing enthalpies in liquid Mg-Si system at 1773 K combined with calculated results and optimized ones [12,13] at 1350 K
According to the cohesive energies listed in Table 1, the calculated mixing enthalpies in liquid Mg-Si system at 1773 K are plotted in Fig. 2. Compatible with the optimized results [12,13] at 1350 K, the calculated mixing enthalpy reaches the minimum on Mg-rich side. However, the deviations between the AIMD results and optimized ones are obvious. Note that 1350 K is lower than the melting point of Si, and as mentioned in Introduction, no direct measurement for the mixing enthalpy is available in liquid Mg-Si system. Hence, it is worth reassessing the reliabilities of the optimized results by means of AIMD [33].
We sampled 20 at.% Si, 40 at.% Si, and 60 at.% Si to reassess the mixing enthalpies at 1350 K. The corresponding physical parameters and PPCFs are given in Table 2 and Fig. 3, respectively. Herein, liquid pure Si was obtained by rapid quenching with the speed of 3.33��1014 K/s from 1773 K and confirmed to be supercooled. The calculated mixing enthalpies at 1350 K are between the AIMD results at 1773 K and the results of ELDRIDGE et al [12] as presented in Fig. 2, which is in agreement with the temperature-dependence of mixing enthalpy following LE rule [34] and favors the argument of YUAN et al [11] that the results of ELDRIDGE et al [12] should be more reliable compared with the results of GEFFICEN and MILLER [13].
To explore the atomic interaction mechanism in the evolution of the mixing enthalpy, we calculated the transferred charges and electron density differences in liquid Mg-Si system. Figure 4 exhibits the calculated average transferred charges per atom, and a dash line is also provided to denote the expected values based on the transferred electron affinity of Si at 6.25 at.% Si. The deviations between the calculated results and dash line suggest that the electrons resist to transfer from Mg towards Si above 18.75 at.% Si. Figures 5(a1) and (a2) show the electron density differences at 18.75 at.% Si. Once Si atoms appear near Mg, the electron density differences become positive between Mg and Si, while the differences are close to zero between Mg atoms. This indicates that the formation of Mg-Si interactions is contributed by the electron redistribution around Mg, which weakens Mg-Mg interactions. At 18.75 at.% Si, the number of Mg atom is much larger than that of Si, which means that Mg atoms can occupy most of positions within the first coordination shell of Si to form Mg-Si interactions. Even so, the positive electron density differences still occur between Si atoms. This result reflects that Si-Si interactions are sufficiently stable to be maintained even on Mg-rich side, and the formation of Mg-Si interactions cannot break up Si-Si interactions.
Table 2 Physical parameters of liquid Mg-Si alloys at 1350 K
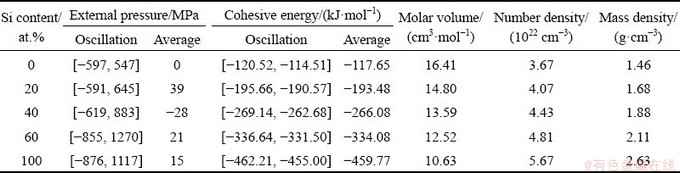
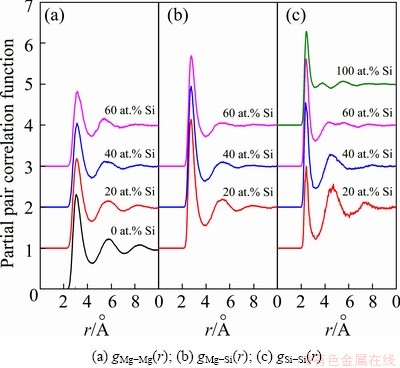
Fig. 3 Partial pair correlation functions of liquid Mg-Si alloys at 1350 K
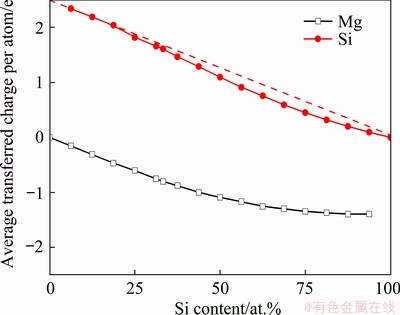
Fig. 4 Calculated average transferred charges per atom in liquid Mg-Si system at 1773 K (The dash line denotes the expected values based on the transferred electron affinity of Si at 6.25 at.% Si)
Moreover, the electron density differences at 50 at.% Si in Figs. 5(b1) and (b2) demonstrate that the chemical environments around Si atoms are dominated by Si-Si interactions despite the abundant Mg-Si interactions, because the electrons around Si prefer aggregating among Si atoms rather than between Mg and Si. Considering that liquid pure Si with only Si-Si interactions has the most negative cohesive energy in the binary system, the dominance of the stable Si-Si interactions determines that the minimum mixing enthalpy is located on Mg-rich side.
3.2 Structural order
As plotted in Fig. 6, the calculated PPCFs in liquid Mg-Si system can be classified into three groups: gMg-Mg(r), gMg-Si(r), and gSi-Si(r). WASEDA [27] measured the PPCF of liquid pure Mg at 1153 K and Si at 1793 K via high energy X-ray diffractometry, which are also provided. By comparison, our calculated PPCF of liquid pure Mg still preserves the character of the liquid with weakened short-range order, and a considerable agreement is reached between the calculated result and experimental one for liquid pure Si. The PPCFs further confirm that the simulated liquid Mg-rich alloys are overheating melts at 1773 K and the AIMD simulations are performed reasonably in this work. On this basis, the partial and total coordination numbers (ZMg-Mg, ZMg-Si, ZSi-Mg, ZSi-Si and ZMg, ZSi) are given in Table 3.
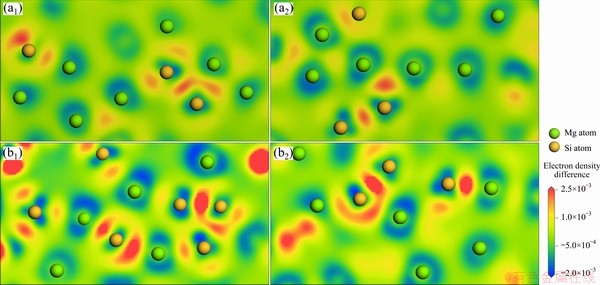
Fig. 5 Electron density differences in liquid Mg195Si45 (a1, a2) and Mg120Si120 (b1, b2) alloys
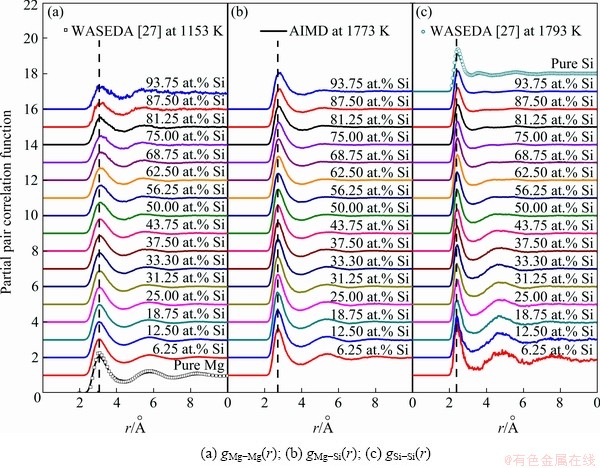
Fig. 6 Partial pair correlation functions in liquid Mg-Si system at 1773 K
Table 3 Partial and total coordination numbers in liquid Mg-Si system at 1773 K
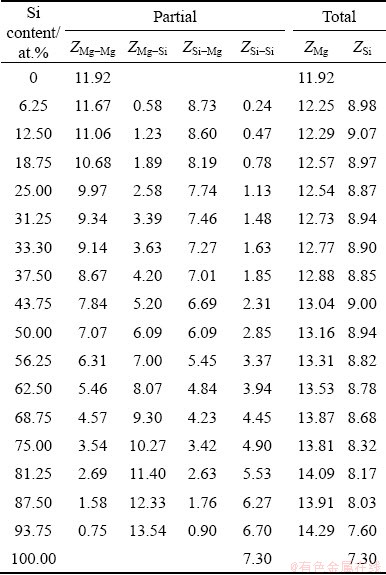
In order to characterize the structural order, we analyzed HA bond pairs [32] within the first peaks of the PPCFs. Figure 7 exhibits the numbers of Type 1 root pairs per 240 atoms in liquid Mg-Si system. Compared with Si-rich side, more Type 1 root pairs are searched out on Mg-rich side because of the larger total coordination numbers. The number of Mg-Si pairs reaches the maximum on Si-rich side, and simultaneously, Si-Si pairs proliferate exponentially to be more numerous than Mg-Mg pairs. Thus, Mg-Si pairs are prone to appear cooperatively with Si-Si pairs rather than Mg-Mg pairs.
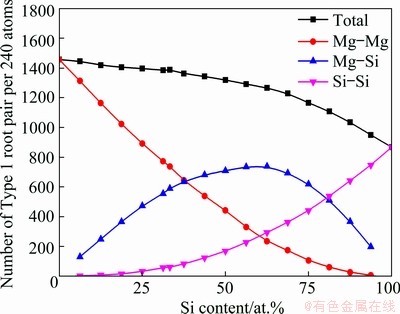
Fig. 7 Numbers of Type 1 root pairs per 240 atoms in liquid Mg-Si system at 1773 K
The number of shared neighbors around a root pair represents the local symmetry of the atomic arrangement. Then the total formation abilities of each local symmetry in liquid Mg-Si system are illustrated by the percentages of Type 1 root pairs with j (j=1, 2, 3, 4, 5, 6) shared neighbors in Fig. 8(a). Except the threefold symmetry that is important on both Mg-rich and Si-rich sides, each of the others presents a monotone variation of the total formation ability dependent on Si content. The fourfold and fivefold symmetries dominate the arrangements of shared neighbors around Type 1 root pairs on Mg-rich side, and their total formation abilities deteriorate with the increase of Si content. On the other hand, the total formation ability of the onefold and twofold symmetries improve to be superior on Si-rich side.
From Figs. 8(b-d), the formation abilities of local symmetries are categorized around Mg-Mg, Mg-Si, and Si-Si pairs, respectively. Interestingly, each symmetry around Mg-Mg pairs has a nearly constant formation ability independent on Si content, and the formation abilities of local symmetries around Si-contained pairs (Mg-Si and Si-Si) result in the variations of the total ones. These results reflect that the addition of shared neighboring Si atoms around Mg-Mg pairs offsets the reduction of Mg with Si content increasing, while the vacancies created by the reduction of shared neighboring Mg atoms around Si-contained pairs are not occupied completely by the additional Si.
According to the defined parameter , the atomic distribution behaviors around Type 1 root pairs are revealed in Fig. 9. For Mg-Mg pairs, the attraction to Mg atoms and repulsion to Si atoms are reversed near 50 at.% Si, which is essential for the fact that the addition of shared neighboring Si atoms can offset the reduction of Mg and vice versa.
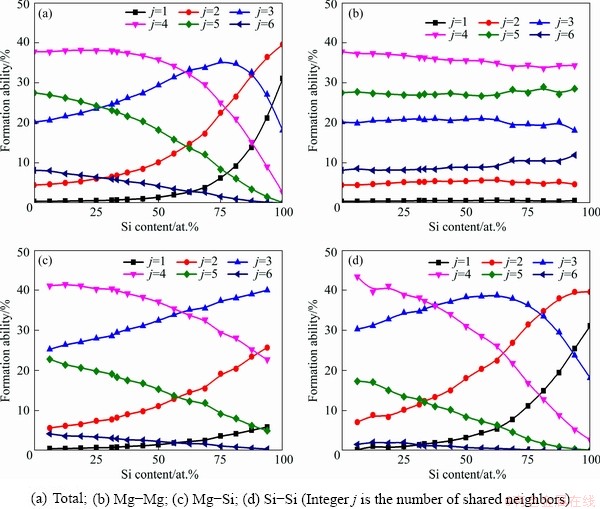
Fig. 8 Formation abilities of local symmetries around Type 1 root pairs in liquid Mg-Si system at 1773 K
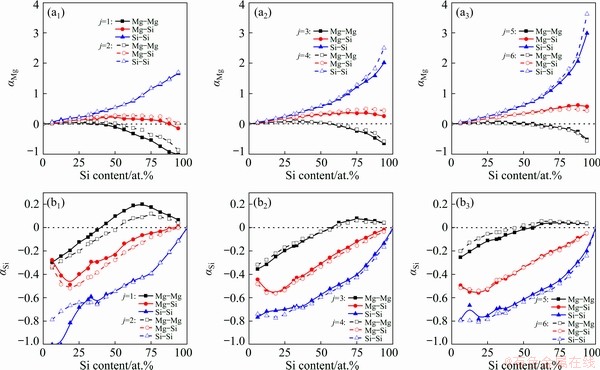
Fig. 9 Atomic distribution behaviors of Mg (a1-a3) and Si (b1-b3) around Mg-Mg, Mg-Si, and Si-Si pairs in liquid Mg-Si system at 1773 K
Si-contained pairs persist attracting Mg atoms and repulsing Si atoms (the only exception is located at 93.75 at.% Si for Mg-Si pairs with 1 shared neighbor). Especially for Si-Si pairs, the higher the local symmetry is, the more significant the attraction and repulsion will be. Such atomic distribution behaviors demonstrate that Mg-Si pairs are prone to appear cooperatively with Si-Si pairs, which is mainly attributed to the attraction between Si-Si pairs and Mg atoms. More importantly, Mg atoms prefer to being arranged in high symmetries while Si atoms in low symmetries. As a consequence, the vacancies created by the reduction of shared neighboring Mg atoms cannot be occupied completely by the additional Si with Si content increasing.
Furthermore, there is an extremum in each series of 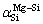 and
and 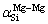 . The repulsion between Mg-Si pairs and Si atoms fades from 18.75 at.% Si, which contributes to the proliferation of Si-Si pairs around Mg atoms. When Si content is up to the region of 68.75-75.00 at.%, the attraction between Mg-Mg pairs and Si atoms turns to be weakened. Combined with the persisting attraction between Si-Si pairs and Mg atoms and the reversals of atomic distribution behaviors around Mg-Mg pairs, the extrema in reflect that the initial aggregated Mg atoms are dispersed among the proliferated Si-Si pairs, and the proliferated Si-Si pairs serve as the framework of the melts.
. The repulsion between Mg-Si pairs and Si atoms fades from 18.75 at.% Si, which contributes to the proliferation of Si-Si pairs around Mg atoms. When Si content is up to the region of 68.75-75.00 at.%, the attraction between Mg-Mg pairs and Si atoms turns to be weakened. Combined with the persisting attraction between Si-Si pairs and Mg atoms and the reversals of atomic distribution behaviors around Mg-Mg pairs, the extrema in reflect that the initial aggregated Mg atoms are dispersed among the proliferated Si-Si pairs, and the proliferated Si-Si pairs serve as the framework of the melts.
HA bond pair is eventually ascertained by counting the number of bonds among the shared neighbors, thereby the average dense degrees of atomic arrangements around Type 1 root pairs can be quantified according to the defined parameter ��root. It can be surmised based on the atomic sizes that the densest arrangements should occur around Si-Si pairs. As exhibited in Fig. 10, such a case is matched for the fourfold and fivefold symmetries. However, for the twofold and threefold symmetries, the average dense degrees around Si-Si pairs are not the highest in the region that even extends to Si-rich side. In view of the fact that all the local symmetries around Si-Si pairs benefit from the attraction to Mg atoms, Si-Si pairs actually play a role in dispersing Mg atoms. What is more, Si addition leads to the reduction of the average dense degrees. Therefore, Si atoms are arranged following low dense degrees in liquid Mg-Si system, which confirms that the proliferated Si-Si pairs constitute frame structures infilled by the dispersed Mg atoms.
3.3 Correlation between mixing enthalpy and structural order
Inspired by the results above, the proliferation of Si-Si pairs might correlate with the evolution of the mixing enthalpy in liquid Mg-Si system. Several studies [35-37] confirm that the tetrahedral atomic arrangement mode is preserved in liquid pure Si, which corresponds to the basic unit of the crystalline structure (diamond structure). Hence, we attempted to seek a correlation between the mixing enthalpy and structural order by focusing on tetrahedral order of local Si-configurations.
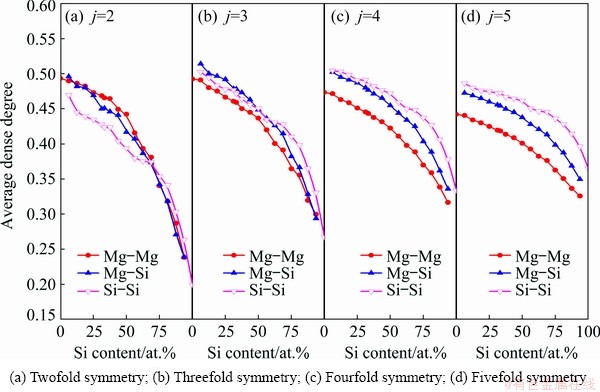
Fig. 10 Average dense degrees of atomic arrangements around Type 1 root pairs in liquid Mg-Si system at 1773 K
The inset of Fig. 11 shows the distribution of tetrahedral order P(qt) in liquid pure Si. A main peak is located at qt=0.5, revealing that the tetrahedral atomic arrangement mode is available mainly in the region 0.5<>t��1. On this basis, the ratio P(0.5<>t��1)/P(qt��1) is utilized to estimate the proportion of the tetrahedral type in local Si-configurations.
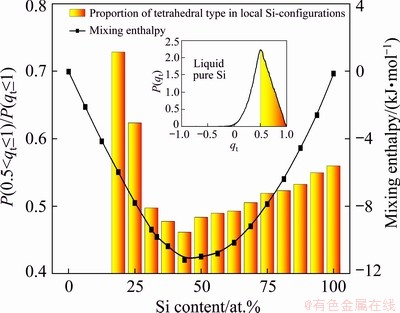
Fig. 11 Correlation between mixing enthalpy and structural order in liquid Mg-Si system at 1773 K (Inset: distribution of tetrahedral order in liquid pure Si)
By plotting the ratio P(0.5<>t��1)/P(qt��1) combined with the mixing enthalpy dependent on Si content, a correlation is shown in Fig. 11, which illustrates that Si-Si interactions are beneficial to the tetrahedral atomic arrangements of Si while Mg-Si interactions have the opposite effect. The tetrahedral type appears from 18.75 at.% Si where the electrons resist to transfer from Mg towards Si initially. Although local Si-configurations are rare at 18.75 at.% Si, the tetrahedral type still has a high proportion owing to the maintained Si-Si interactions. Further addition of Si enriches local Si-configurations and Mg-Si interactions, and consequently, both the proportion of the tetrahedral type and mixing enthalpy evolve to be minimum. Nevertheless, the dominance of Si-Si interactions in the chemical environments around Si makes the proportion of the tetrahedral type turn to rise accompanied with the mixing enthalpy on Mg-rich side.
4 Conclusions
(1) The calculated mixing enthalpies in liquid Mg-Si system have reasonable agreement with the optimized ones based on isopiestic experiments. The minimum is located on Mg-rich side, which is determined by the fact that Si-Si interactions dominate the chemical environments around Si.
(2) For the structural order, the reversals of atomic distribution behaviors occur around Mg-Mg pairs near 50 at.% Si, which is essential for keeping the formation abilities of local symmetries independent of Si content; Si-contained pairs persist attracting Mg atoms and repulsing Si, which contributes to the facts that Mg-Si pairs are prone to appear cooperatively with Si-Si pairs and the formation ability of local symmetries around Si-contained pairs varies with Si content.
(3) The evolution of structural order with Si content increasing can be characterized as follows: the initial aggregated Mg atoms are gradually dispersed by Si-Si pairs, and the proliferated Si-Si pairs constitute frame structures infilled by the dispersed Mg atoms on Si-rich side.
(4) The uncovered correlation between the mixing enthalpy and structural order illustrates that Si-Si interactions are beneficial to the tetrahedral ordered arrangements of Si atoms while Mg-Si interactions have the opposite effect.
Acknowledgments
The authors are grateful for the financial supports from the National Key Research and Development Program of China (2016YFB0701202), the National Natural Science Foundation of China (51901117, 51801116, 51804190, and 11804179), and the Shandong Provincial Key Research and Development Plan, China (2019GGX102047).
References
[1] QIN Qing-dong, ZHAO Hong-long, LI Juan, ZHANG Ying-zhe, SU Xiang-dong. Microstructure and mechanical properties of friction stir processed Al-Mg2Si alloys [J]. Transactions of Nonferrous Metals Society of China, 2020, 30: 2355-2368.
[2] WANG Wei-dan, GAO Ming, HUANG Yuan-ding, TAN Li-li, YANG Ke, HORT N. Microstructures, corrosion and mechanical properties of Mg�CSi alloys as biodegradable implant materials [C]//Proc Magnesium Technology 2019. San Antonio, TX: TMS, 2019: 151-157.
[3] ZHOU Xiong, GUO Tian, WU Shu-sen, LU Shu-lin, YANG Xiong, GUO Wei. Effects of Si content and Ca addition on thermal conductivity of as-cast Mg-Si alloys [J]. Materials, 2018, 11: 2376.
[4] VOGEL R. On the magnesium-silicon alloys [J]. Zeitschrift fur Anorganische Chemie, 1909, 61: 46-53.
[5] DORNER P, KRIEG H, LUKAS H L, M��LLER R, PELZOW G. The system Mg�CSi optimized by a least squares method [J]. Calphad, 1981, 1: 41-55.
[6] L��DECKE D. Phase diagram and thermochemistry of the Al-Mg-Si system [J]. Zeitschrift fur Metallkunde, 1986, 77: 278-283.
[7] CHAKRABORTI N, LUKAS H L. Thermodynamic optimization of the Mg-Al-Si phase diagram [J]. Calphad, 1992, 16: 79-86.
[8] FEUFEL H, GODECKE T, LUKAS H L, SOMMER F. Investigation of the AlMgSi system by experiments and thermodynamic calculations [J]. Journal of Alloys and Compounds, 1997, 247: 31-42.
[9] YAN X Y, CHANG Y A, ZHANG F. A thermodynamic analysis of the Mg-Si system [J]. Journal of Phase Equilibria, 2000, 21: 379-384.
[10] JUNG I H, KANG D H, PARK W J, KIM N J, AHN S H. Thermodynamic modeling of the Mg-Si-Sn system [J]. Calphad, 2007, 31: 192-200.
[11] YUAN Xiao-ming, SUN Wei-hua, DU Yong, ZHAO Dong-dong, YANG Hua-ming. Thermodynamic modeling of the Mg�CSi system with the Kaptay equation for the excess Gibbs energy of the liquid phase [J]. Calphad, 2009, 33: 673-678.
[12] ELDRIDGE J M, MILLER E, KOMAREK K L. Thermodynamic properties of liquid magnesium�Csilicon alloys: Discussion of the Mg-group IVB systems [J]. Transactions of the Metallurgical Society of AIME, 1967, 239: 775-781.
[13] GEFFICEN R, MILLER E. Phase diagrams and thermodynamic properties of the Mg-Si and Mg-Ge systems [J]. Transactions of the Metallurgical Society of AIME, 1968, 242: 2323-2328.
[14] KAPTAY G. The exponential excess Gibbs energy model revisited [J]. Calphad, 2017, 56: 169-184.
[15] GHASEMIA M, STUTZ E, STEINVALL S E, ZAMANI M, MORRAL A F. Thermodynamic re-assessment of the Zn-P binary system [J]. Materialia, 2019, 6: 100301.
[16] PELTON A D, DECTEROV S A, ERIKSSON G, ROBELIN C, DESSUREAULT Y. The modified quasichemical model I-Binary solutions [J]. Metallurgical and Materials Transactions B, 2000, 31: 651-659.
[17] ISOMAKI I, ZHANG R, XIA L G, HELLSTEN N, TASKINEN P A. Thermodynamic assessment of ZnO-SiO2 system [J]. Transactions of Nonferrous Metals Society of China, 2018, 28: 1869-1877.
[18] LIU Dan, ZHU Xun-ming, QIN Jing-yu, WANG Ai-min, DUAN Jun-peng, GU Ting-kun. First-principles study of chemical and topological short-range orders in the Mg-Si liquid alloys [J]. Metals, 2016, 6: 78.
[19] QIN Jing-yu, LI Xin-xin, WANG Jin, PAN Shao-peng. The self-diffusion coefficients of liquid binary M-Si (M= Al, Fe, Mg and Au) alloy systems by first principles molecular dynamics simulation [J]. AIP Advances, 2019, 9: 035328.
[20] KRESSE G, FURTHM��LLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Computational Materials Science, 1996, 6: 15-50.
[21] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Physical Review B, 1999, 59: 1758-1775.
[22] PERDEW J P, CHEVARY J A, VOSKO S H, JACKSON K A, PEDERSON M R, SINGH D J, FIOLHAIS C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation [J]. Physical Review B, 1992, 46: 6671.
[23] PERDEW J P, WANG Y. Accurate and simple analytic representation of the electron-gas correlation energy [J]. Physical Review B, 1992, 45: 13244-13249.
[24] NOSE S. A unified formulation of the constant temperature molecular dynamics methods [J]. The Journal of Chemical Physics, 1984, 81: 511-519.
[25] HOOVER W G. Canonical dynamics: Equilibrium phase- space distributions [J]. Physical Review A, 1985, 31: 1695-1697.
[26] SWOPE W C, ANDERSEN H C, BERENS P H, WILSON K R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters [J]. The Journal of Chemical Physics, 1982, 76: 637-649.
[27] WASEDA Y. The structure of non-crystalline materials [M]. New York: McGraw-Hill, 1980.
[28] TANG W J, SANVILLE E, HENKELMAN G. A grid-based Bader analysis algorithm without lattice bias [J]. Journal of Physics: Condensed Matter, 2009, 21: 084204.
[29] WANG Jin, LI Xin-xin, PAN Shao-peng, QIN Jing-yu. Mg fragments and Al bonded networks in liquid Mg-Al alloys [J]. Computational Materials Science, 2017, 129: 115-122.
[30] HONEYCUTT J D, ANDERSEN H C. Molecular dynamics study of melting and freezing of small Lennard-Jones clusters [J]. The Journal of Chemical Physics, 1987, 91: 4950-4963.
[31] WARREN B E, AVERBACH B L, ROBERTS B W. Atomic size effect in the X-ray scattering by alloys [J]. Journal of Applied Physics, 1951, 22: 1493-1496.
[32] ERRINGTON J R, DEBENEDETTI P G. Relationship between structural order and the anomalies of liquid water [J]. Nature, 2001, 409: 318-321.
[33] WANG Jin, ZHANG Qiang, QIN Jing-yu. Structural transition region of liquid Mg�CLi alloys [J]. Computational Materials Science, 2016, 117: 259-265.
[34] KAPTAY G. On the tendency of solutions to tend toward ideal solutions at high temperatures [J]. Metallurgical and Materials Transactions A, 2012, 43: 531-543.
[35] GANESH P, WIDOM M. Liquid-liquid transition in supercooled silicon determined by first-principles simulation [J]. Physical Review Letters, 2009, 102: 075701.
[36] VASISHT V V, SAW S, SASTRY S. Liquid-liquid critical point in supercooled silicon [J]. Nature Physics, 2011, 7: 549-553.
[37] HE Y X, LI J S, WANG J, BEAUGNON E. Liquid-liquid structure transition in metallic melt and its impact on solidification: A review [J]. Transactions of Nonferrous Metals Society of China, 2020, 30: 2293-2310.
Һ̬Mg-Siϵ�л������ṹ��֮��������
�� ��1���ؾ���2���ܼ�ѧ1���̿���1��ղ��ΰ1��������1���Թ���1��������3����˳�4���� ��5
1. ��³��ҵ��ѧ(ɽ��ʡ��ѧԺ) ɽ��ʡ��ѧԺ �²����о��� ɽ��ʡ���ʸ�ǿ���������ص�ʵ���ң����� 250014��
2. ɽ����ѧ ����Һ�̽ṹ�ݱ���ӹ��������ص�ʵ���ң����� 250061��
3. �ijǴ�ѧ ���Ͽ�ѧ�빤��ѧԺ���ij� 252000��
4. ³����ѧ εɽ�����뺣��ѧԺ����̨ 264025��
5. �ൺ�Ƽ���ѧ ����ѧԺ���ൺ 266061
ժ Ҫ�����õ�һ��ԭ�����Ӷ���ѧ�о�1773 K��Һ̬Mg-Siϵ�Ļ������ṹ��ͨ������ת�Ƶ�ɺͲ�ֵ����ܶȣ�֤ʵSiԭ����Χ�Ļ�ѧ������Si-Si��������������ɴ˾���������ڸ�Mg��ﵽ��Сֵ������ƫż��غ�����HA���Է��������ʾ��Si-Siԭ�Ӷ���Mgԭ�����������ҽṹ����Si�������ݱ��������ΪSi-Siԭ�ӶԷ�ɢMgԭ���Թ��ɿ�ܽṹ�Ĺ��̡��Ծ���Siԭ�ӹ��͵���������Ϊ�о�����ʾ�������ṹ��֮�������ԣ�����Ϊ����Һ̬Mg-Si��Ԫϵ�ṩ�ۺ�����ѧ�뼸��ѧ�����ӽǡ�
�ؼ��ʣ�Һ̬Mg-Siϵ������ʣ��ṹ��һ��ԭ�����Ӷ���ѧ
(Edited by Wei-ping CHEN)
Corresponding author: Ji-xue ZHOU, Tel: +86-531-85599042, E-mail: zhoujx@sdas.org;
Jin WANG, Tel: +86-531-88728100, E-mail: wangjin@sdas.org
DOI: 10.1016/S1003-6326(21)65544-9
1003-6326/ 2021 The Nonferrous Metals Society of China. Published by Elsevier Ltd & Science Press
2021 The Nonferrous Metals Society of China. Published by Elsevier Ltd & Science Press
Abstract: The mixing enthalpies and structural order in liquid Mg-Si system were investigated via ab-initio molecular dynamics at 1773 K. By calculating the transferred charges and electron density differences, the dominance of Si-Si interactions in the chemical environments around Si was demonstrated, which determined that the mixing enthalpy reached the minimum on Mg-rich side. In terms of Honeycutt and Anderson (HA) bond pairs based on the partial pair correlation functions, the attraction between Si-Si pairs and Mg atoms was revealed, and the evolution of structural order with Si content was characterized as a process of constituting frame structures by Si-Si pairs that dispersed Mg atoms. Focusing on tetrahedral order of local Si-configurations, a correlation between the mixing enthalpy and structural order was uncovered ultimately, which provided a new perspective combining the energetics with geometry to understand the liquid Mg-Si binary system.
