网络首发时间: 2018-07-23 18:30
稀有金属 2019,43(06),631-637 DOI:10.13373/j.cnki.cjrm.xy18040039
锑空位复合体对锗晶体物性影响的第一性研究
杨晓京 耿瑞文 谢启明 罗良 李芮
昆明理工大学机电工程学院
云南北方驰宏光电有限公司
昆明理工大学环境科学与工程学院
摘 要:
利用第一性原理软件CASTEP系统研究了锑掺杂锗晶体中锑原子与空位的相互作用, 对单空位、双空位可能出现的结构模型进行了计算, 通过对结合能的比较分析了相应的结构模型的稳定性。结果表明, 在锑掺杂锗晶体中由于锑的引入, 空位倾向于聚集在锑原子周围, 形成锑-空位复合体。当体系仅含有一个空位时, 空位处于锑原子的第一近邻晶格位点时形成最稳定的结构其结合能为-1.10 eV。当体系含有两个空位时, 双空位分别位于锑原子同一共价键方向上的第一、第二近邻晶格位点形成最稳定的结构其结合能为-2.03 eV。使用Voigt-Reuss-Hill近似计算相应单空位、双空位模型的弹性模量并与无缺陷锗晶体的弹性模量相比较, 根据Pugy判据可知无缺陷锗晶体的B /G (体模量/剪切模量) 值为1.38, 由于锑空位复合体的引入使单空位双空位模型的B /G 值增加到1.59~1.73的范围之内, 由此表明锑空位复合体可以提高锗晶体的塑性, 这也与利用柯西压力判定脆塑性得到的结论一致。
关键词:
第一性原理 ;锗晶体 ;锑空位复合体 ;稳定性 ;塑性 ;
中图分类号: O614.431;O731
作者简介: 杨晓京 (1971-) , 男, 云南大理人, 博士, 教授, 研究方向:红外光学材料的超精密加工及材料计算;电话:13668718025;E-mail:xjyang@vip.sina.com;
收稿日期: 2018-04-19
基金: 国家自然科学基金项目 (51765027); 昆明理工大学分析测试基金项目 (2018P20173103002) 资助;
Physical Properties of Germanium Crystal with Antimony Vacancy Complexes by First-Principle Investigation Yang Xiaojin Geng Ruiwen Xie Qiming Luo liang Li Rui
Faculty of Mechanical and Electrical Engineering, Kunming University of Science and Technology
Yunnan KIRO-CH Photonics Co., Ltd.
Faculty of Environmental Science and Engineering, Kunming University of Science and Technology
Abstract:
The first-principle theory software (CASTEP) was used to systematically study interaction between antimony and vacancies in germanium crystal, the possible structure models of monovacancy and pacancy were calculated, and the stability of related models were analyzed by comparing the binding energy. As the results showed, because of the introducing of antimony atom, vacancies tended to gather around with antimony atom, and formed antimony-vacancy complex in Ge crystal. For monovacancy case, the most stable structure was formed when the vacancy was located in the first nearest-neighboring of antimony atom, and the binding energy was-1.10 eV. For pacancy case, the most stable structure was formed when the pacancy was respectively located in the first and second nearest-neighboring of the same covalent bond branch of antimony atom, and the binding energy was-2.02 eV. Voigt-Reuss-Hill approximation was used to calculate elastic modulus of monovacancy, pacancy and defect free case in Sb doped Ge crystal, and the related elastic modulus were compared. According to Pugy criterion, the B /G (bulk modulus/shear modulus) value increased from 1.38 for defect free case to a range of 1.59~1.73 for monovacancy and pacancy case, and the results showed that the antimony-vacancy complex could improve the plasticity of Ge crystal. This was also consistent with the conclusion obtained by using the Cauchy pressure to determine brittle and plasticity.
Keyword:
first principles; germanium crystal; antimony vacancy complex; stability; plasticity;
Received: 2018-04-19
锗是重要的半导体材料, n型锗晶体 (掺Sb, P, As等Ⅴ族原子) 具有优异的红外透过性、 高折射率、 低色散性等特点, 常用于红外光学装置的窗口、 透镜与棱镜材料。 锗与硅相比具有比更小的带隙, 较大的载流子迁移率, 更佳的掺杂剂溶解度
[1 ]
, 被视为CMOS的未来沟道材料
[2 ]
, 在先进的纳米电子器件制造领域有望取代硅材料
[3 ]
, 从而重新引起研究者极大的兴趣。 锑掺杂缺陷是在锗晶体中常见的缺陷 (E-center缺陷的一种) , 即在锗晶格中由一个锑原子替换一个锗原子, 掺杂可以影响和控制晶体的微缺陷行为, 因此利用掺杂来控制锗晶体中的缺陷也成为一个重要的研究方向。
Chroneos等
[4 ]
使用密度泛函理论建模研究发现, 在锡磷共掺杂锗晶体中锡原子是空位的有效陷阱。 Markevich等
[5 ]
使用深能级瞬态谱研究n型锗晶体中辐照缺陷发现, 掺杂缺陷的热稳定性随着Ⅴ族原子P, As, Sb, Bi半径的增加而增加。 现有的理论
[3 ,6 ]
与实验
[7 ]
证明掺杂供体原子磷、 砷、 锑在锗晶体中通过与空位的相互作用而扩散, 由于空位的形成能低于自间隙形成能, 在锗晶体中空位缺陷占据主导作用
[8 ]
。 Chen等
[9 ]
研究了直拉晶体硅中锗掺杂缺陷与空位缺陷的相互作用, 使用第一性原理得出在硅晶体生长冷却过程中空位倾向于与锗掺杂原子相互作用, 形成锗空位复合体。
锗作为一种典型的硬脆性材料, 常使用单点金刚石切削技术进行加工, 即可在塑性域实现纳米精度的切削
[10 ]
, 现阶段锗晶体通常由直拉法制备, 其内部不可避免的分布着各种缺陷。 空位与掺杂作为常见的点缺陷, 广泛的存在于锗材料中。 在近年来的研究中, 空位缺陷日益引起研究者的关注。 Fujita等研究发现
[11 ]
当纳米金属晶体在产生高速塑性变形时存在一定数量的空位或者空位团, 但是在此过程中空位的分布特征以及演变方式, 目前尚无实验研究报道。 朱攀攀等
[12 ]
在研究点缺陷对B2-CoSc力学性质的影响时发现, 空位缺陷的存在使材料塑性得到提高。 点缺陷会引起局部的应力与应变, 进而对材料的物理性质和力学性质产生影响, 虽然锑掺杂锗晶体被广泛地应用于红外光学材料, 但关于锗中空位与锑的相互作用缺乏相关的实验研究, 尤其缺乏锗晶体中点缺陷对于力学性质的影响的研究。 正是如此, 研究点缺陷对锗晶体力学性质的影响也显得尤为重要, 但仅仅通过总能量的大小的变化来判定模型的稳定性, 并不具备完全的充分性。 因此本文使用第一性原理在超精细精度下建立了n型锗晶体中锑掺杂缺陷杂与空位的相互作用的物理模型, 通过计算缺陷的结合能, 研究最稳定的缺陷模型, 并通过计算相应模型的弹性常数, 分析缺陷对力学性能的影响规律。
1 模型参数及计算方法
本文使用CASTEP
[13 ]
模块对模型进行几何优优化, 基于密度泛函理论 (density functional theory, DFT) , 采用局域密度近似 (local density approximation, LDA) 下的CAPZ 交换关联泛函进行计算, 在计算过程中, 为了获得精确的结构, 模型参数通过增加截断能和K 点数进行调整, 经过充分验证以保证收敛。 其中平面波基截断能设置为400 eV, 采用非保守赝势 (nor-conserving) , 布里渊区使用Monkhorst-Pack方法, k 点设置为2×2×2, 在Ultra-Fine精度下, 采用BFGS算法
[14 ]
对超胞模型进行优化, 获得稳定的结构模型。 收敛判据如下: Hellmann-Feynaman力不大于0.1 eV・nm-1 ; 最大应力不超过0.02 GPa; 最大区取代位置小于5×10-5 nm; 能量变化小于5×10-6 eV・atom-1 。
锗晶体具有金刚石结构, 其空间点群为FD-3M (227) , Singh
[15 ]
通过实验测得锗晶体在不同温度下的晶格常数并进行3次拟合获得晶格常数与温度的关系函数, 经计算锗晶体在0 K时晶格常数为0.562 nm, 经计算其晶格常数为0.559 nm与实验值相比, 误差仅为1.2%。 Mattsson
[16 ]
发现可以忽略超胞模型尺寸的不同对点缺陷计算结果的影响, 因此本文采用由2×2×2个锗晶胞, 共计64个原子构成的超胞模型, 并用一个锑原子替换超胞中心的锗原子, 建立锑掺杂锗晶体模型, 该模型被广泛地
[9 ,17 ,18 ]
用于掺杂原子对于半导体材料的物性影响的计算。 此时锑原子在超胞中的含量为1.56%, 超胞的晶格长度为1.118 nm。
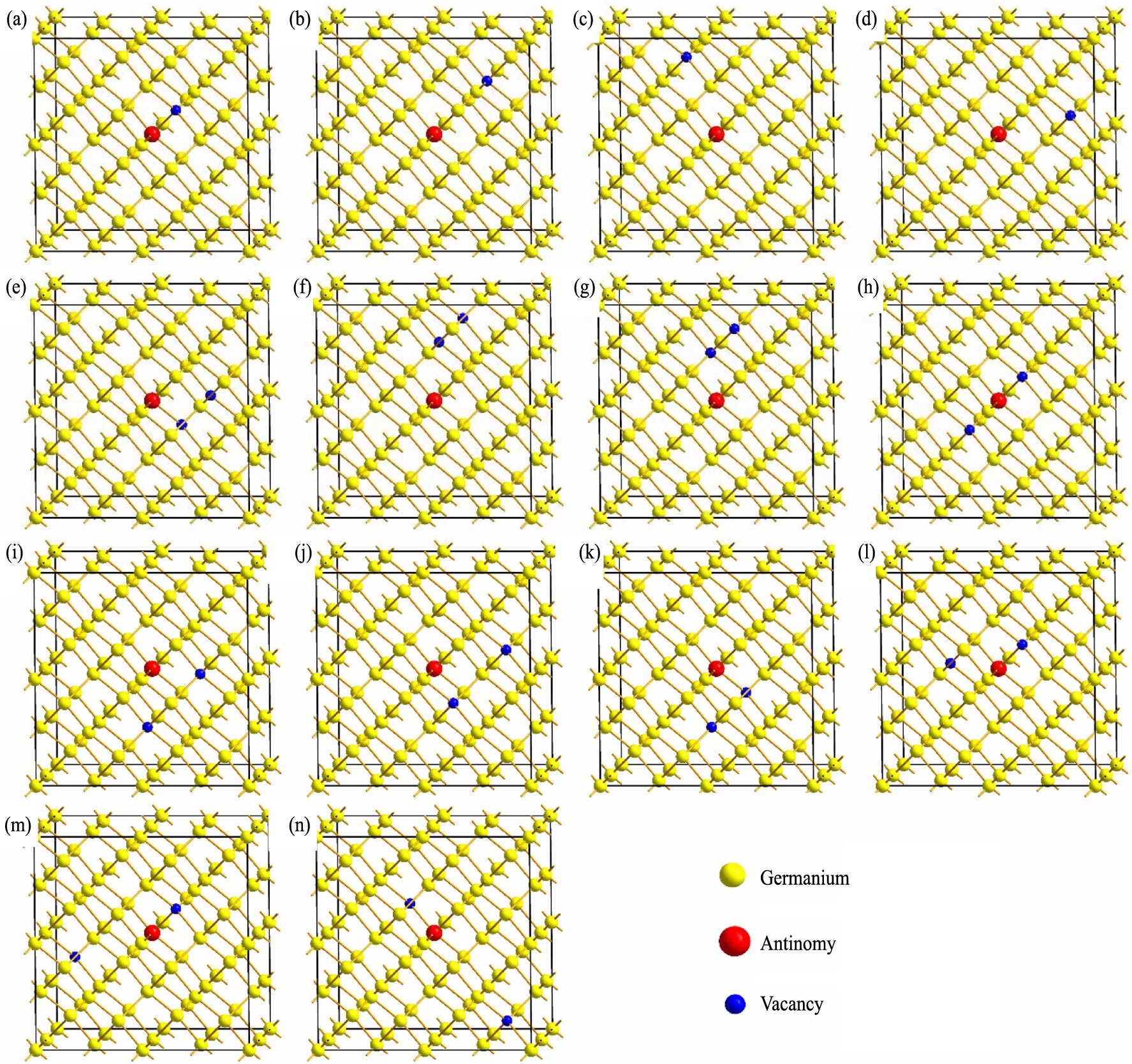
为了研究在锑掺杂锗晶体中锑与空位的相互作用模型中的的位置, 对于空位使用与锑、 锗原子不同颜色的原子替代, 如图1所示。
通过对构建的锑空位复合体模型进行几何优化, 得到了各模型的基态总能量并通过与每组模型最小基态能量比较计算了相对能量。 结果如表1所示, 其中A, B代表不同的共价键方向, 数字i (i =1, 2, 3, 4, 5) 代表锑原子的第i 近邻晶格位点。
图1 锗晶体超胞中锑空位复合体结构模型
Fig.1 Structure models of antinomy-vacancy complex in Ge crystal supercell
表1 锗晶体中锑空位复合体的总能量及相对能量
Table 1 Total energy and relative energy of Sb-Vacancy defect pairs in Ge lattice supercell
Group
Model
Vacancy
Total energyE /eV)
Relative energyE r /eV)
a
A1
-6825.59365
0
b
A2
-6825.27698
0.3167
c
A3
-6825.07713
0.5165
d
A4
-6824.20499
1.3887
e
A1A2
-6716.37768
0
f
A2A3
-6716.10351
0.2742
g
A2A4
-6716.04261
0.3351
h
A1B1
-6715.75031
0.6274
i
A2B2
-6715.52956
0.8481
j
A3A4
-6715.35509
1.0226
k
A1A2
-6716.37198
0.0057
l
A1B2
-6715.65175
0.7259
m
A1B3
-6715.48287
0.8948
n
A1B5
-6715.00308
1.3746
2 结果与讨论
2.1 锑空位复合体结合能与形成能
缺陷间的相互吸引作用可以用结合能 (binding energy) 来描述, 即孤立原子结合成稳定晶体所释放的能量, 当结合能为正数时意味着该缺陷并不可以稳定存在, 通过方程 (1) 定义结合能
[4 ,17 ,19 ]
的计算方式:
E binding =E defect-comple -∑comp E isolated-defect (1)
式中E defect-comple 表示缺陷复合体能量与无缺陷超胞的差值, E isolated-defect 表示仅含一种缺陷的能量与无缺陷锗晶体超胞的差值, 根据定义, 若计算的结合能为负值, 则表示缺陷复合体相对于其孤立缺陷是稳定的。 基于方程 (1) 可以计算锑掺杂锗晶体中单空位的结合能如式 (2) 所示:
E b (SbVGeN -2E (SbVGeN -2E (SbGeN -1E (VGeN -1E (GeN
式中E (SbVGeN -2E (SbGeN -1E (VGeN -1E (GeN N 表示超胞中包含的原子总数, 在本文中N 为64。
在锑掺杂锗晶体中锑空位形成能是表征空位与锑原子形成缺陷复合体的难易程度的一个物理量, 表示在锑掺杂体系里形成锑空位复合体引起的体系总势能的增加。 如果形成能为正值, 表示形成缺陷复合体需要从外界吸收能量, 反之则表示形成缺陷复合体会释放能量即缺陷复合体可只发形成, 用式 (3) 计算单空位锑掺杂锗晶体形成能
[20 ]
:
E f ( S b V 2 G e Ν - 2 ) = E ( S b V G e Ν - 2 ) - E ( S b V G e Ν - 1 ) + 1 2 E ( G e Ν ) ? ? ? ? ? ? ? ? ? ( 3 )
E f ( S b V 2 G e N ? 2 ) = E ( S b V G e N ? 2 ) ? E ( S b V G e N ? 1 ) + 1 2 E ( G e N ) ? ? ? ? ? ? ? ? ? ( 3 )
式中E f (SbV2 GeN -3
首先分析最简单的情况, 即单空位与锑原子的相互作用, 这时空位可能位于锑原子的第一、 第二、 第三近邻等位置, 经过计算得到了不同结构的锑空位复合体的结合能与形成能, 如表2所示。
表2 锑单空位复合体结合能、 形成能与锑原子距离
Table 2 Binding energy (E b formation energy (E f and Sb-V spacing in Sb-V pairs
Binding energyE b /eV)
Formation energyE f /eV)
Sb-V spacing/-10 m
-1.1053
1.3476
2.45
-0.7886
1.6642
4.05
-0.5888
1.8641
6.16
0.2833
2.7362
6.96
计算表明随着空位与锑原子距离的增加, 空位缺陷的形成能逐渐升高, 这意味着单空位更倾向于聚集在锑原子的第一近邻, 以保持结构的稳定性, 而模型d的结合能为负值, 这表明该缺陷可能不会稳定存在。 这也与表1中基态总能量的变化规律一致, 即模型a, b, c, d的总能量依次上升, 这也意味着, 模型结构的稳定性依次下降。 由此可以得出, 当体系只含有一个空位时, 空位倾向于聚集在锑原子所在的晶格位置的第一近邻位点并形成锑空位复合体。
在本文中考虑双空位分别与锑原子发生相互作用, 双空位结合能可由式 (4) 计算, 计算结果如表3所示。
E b (SbV2 GeN -2E (SbV2 GeN -3E (SbGeN -1E (VGeN -1E (GeN
式中E b (SbV2 GeN -3
锑掺杂锗晶体中锑双空位缺陷复合体的形成能, 表示为在锑掺杂锗晶体中引入双空位所需的能量, 其计算方式如式 (5) 所示:
E f ( S b V 2 G e Ν - 3 ) = E ( S b V 2 G e Ν - 3 ) - E ( S b G e Ν - 1 ) + 2 Ν E ( G e Ν ) ? ? ? ? ? ? ? ? ? ( 5 )
E f ( S b V 2 G e N ? 3 ) = E ( S b V 2 G e N ? 3 ) ? E ( S b G e N ? 1 ) + 2 N E ( G e N ) ? ? ? ? ? ? ? ? ? ( 5 )
式中E f (SbV2 GeN -3
当考虑锑掺杂锗晶体模型中存在双空位时, 空间模型会有很多种, 其存在形式可以分为如下3种情况, 第一是两个空位分别位于相邻的两个晶格位点, 即空位对之间间距为0.245 nm。 第二是两个空位分别位于次相邻的位点, 即空位对之间的间距为0.400 nm。 第3种情况是两个空位不相邻, 即空位对之间的距离大于等于0.469 nm。 计算结果如表3所示。
结合表3发现当双空位处于相邻的晶格位点时, 此时两空位之间距离为0.245 nm, 即模型e, f, g, 随着空位几何中心与锑原子间距离的增加, 从0.308~0.505 nm, 缺陷复合体的结合能逐渐减少, 形成能逐渐增加, 这意味着模型e是最稳定结构, 空位对倾向于聚集到与锑原子处于同一共价键方向上的第一近邻、 第二近邻的晶格位点。
表3 双空位锑缺陷复合体的结合能、 形成能、 空位间距与锑原子与空位对中心间距
Table 3 Binding energy, formation energy, V-V spacing and Sb-V spacing of Sb-pacancy defect
e
f
g
h
i
j
k
l
m
n
-2.03
-1.76
-1.69
-1.40
-1.18
-1.00
-2.02
-1.30
-1.13
-0.65
2.88
3.15
3.22
3.51
3.73
3.90
2.87
3.61
3.77
4.25
-10 m) 2.45
2.45
2.45
4.00
4.00
4.00
4.69
6.17
6.93
9.80
-10 m) 3.08
4.18
5.05
1.41
3.46
5.10
2.35
1.23
1.41
2.45
当考虑双空位处于次相邻晶格位点时, 即两空位之间的距离为0.400 nm, 如模型h, i, j所示, 随着空位对中心与锑原子的距离逐渐从0.141 nm增大到0.510 nm, 缺陷复合体的形成能逐渐减小, 这也意味着模型结构稳定性的下降, 同时形成能由3.51 eV逐渐升高至3.90 eV, 这也意味着缺陷形成能力的下降, 即空位对中心与锑原子间距越小, 系统越稳定且越容易形成。 这也与文中单空位的稳定性结论一致。
综合模型j, k, l来看, 空位距离增加的同时, 空位中心与锑原子逐渐减小, 而双空位的结合能却在逐渐增大, 这表明空位对的间距对于系统稳定性的影响大于空位对中心与锑原子间距的影响, 空位对的间距越小, 系统越稳定。 这种规律也适用于双空位缺陷晶格位点不相邻的情形, 如模型k, l, m, n所示, 双空位的距离依次逐渐从0.469 nm增加到0.980 nm, 在这4组模型中, 结构k具备最低的结合能, 虽然模型l与模型k相比其空位对中心与锑原子距离更近 (分别为0.122和0.235 nm) , 但是由于模型k空位对间距为0.469 nm大于模型l的0.617 nm, 模型k拥有比模型l更低的结合能。 这说明随着空位对距离的增加, 超胞体系的能量上升, 复合缺陷的结合能增大, 体系将会变得不稳定, 表明在锗晶体中空位倾向于聚集, 以维持体系稳定。 综合组二的十组数据分析, 模型e拥有最低的结合能其大小为-2.03 eV, 且最容易形成其结合能大小为2.88 eV, 表明双空位缺陷倾向于扩散到锑原子的同一等价共价键的第一近邻和第二近邻晶格位点。
2.2 缺陷对于结构力学性质的影响
空位是固体的一种常见的缺陷, 为了获得材料的力学性质, 研究不同结构锑空位复合体对于锗晶体力学特性的影响就显得尤为重要。 弹性常数是描述材料对外应力响应的重要参数, 锗的弹性常数可以很好地反映其弹性特征, 对于立方晶体而言, 只需计算出C 11 , C 12 , C 44 这3个弹性刚度系数即可。 经过计算得到了锗的晶格常数和二阶弹性常数如表4所示, 与文献值和实验值相比差异很小, 这也充分说明了计算的合理性。 本文分别计算了具有良好稳定性的包含单空位与双空位缺陷的4组结构模型。 采用Voigt-Reuss-Hill近似来描述弹性常数
[21 ]
, 即对在晶粒边界上分别基于连续应力连续性计算的G v 和应变连续的G R 取平均值, 计算公式如下:
B = (C 11 +2C 12 ) /2 (6)
G V = (C 11 -C 12 +3C 44 ) /5 (7)
G R =5C 44 (C 11 -C 12 ) / (4C 44 +3C 11 -3C 12 ) (8)
G = (G R +G V ) /2 (9)
E =9GB / (3B +G ) (10)
杨氏模量描述了材料应力与应变之间的线性响应, 杨氏模量越大, 材料的刚度越大, 其值越小, 材料的塑性越好
[22 ]
, 可由式 (8) 求得
[23 ,24 ]
, 随着锑掺杂与空位的引入, 含有缺陷的锗晶体的弹性模量相较于无缺陷锗晶体而言逐渐降低, 这也意味着材料塑性的逐渐增强, 计算结果如表5所示。
表4 锗的平衡晶格常数与二阶弹性刚度实验值与计算值
Table 4 Calculated and experiment equilibrium lattice parameters and the second order elastic stiffness of Ge
Parameters
This work
Previous
Calculations
Experiments
Ge
LDA
GGA
PW-PP
A /10-10 m5.59
5.763[27]
5.65[16]
C 11 /GPa133.2
103.1[27]
128.5[28]
128.4[29]
129[30]
C 12 /GPa49.7
36.8[27]
45.7[28]
48.23[29]
47.9[30]
C 44 /GPa68.6
53.1[27]
66.8[28]
66.66[29]
67.0[30]
表5 锑空位复合体体模量 (B) 、 剪切模量 (G) 、 杨氏模量 (E) 、 泊松比 (ν) 及柯西压力 (C12-C44)
Table 5 Bulk modulus (B ) , shear modulus (G ) , Young′s modulus (E ) , poisson ratio (ν ) and Cauchy pressure (C 12 C 44 of Sb-vacancy pairs in Ge supercell
Ge
SbV2
b
e
k
ν 0.21
0.24
0.25
0.25
0.24
E /GPa135.81
110.60
108.87
95.39
101.44
B /GPa77.49
70.94
71.52
65.65
67.26
G /GPa56.21
44.59
43.68
37.92
40.62
B /G 1.38
1.59
1.64
1.73
1.66
C 12 -C 44 ) /GPa-18.97
-8.06
-5.72
-3.02
-2.90
根据Pugh关于材料的塑性与脆性判据
[25 ]
, 若B /G 大于1.75, 则材料表现出塑性, 反之材料则为脆性, 无缺陷锗晶体的B /G 值为1.38, 这说明其具有本征塑性, 随着空位的引入使得其塑性得到加强, 对于最稳定的单空位模型 (a和b) 和双空位 (e和k) 模型而言, B /G 值分别在1.59, 1.64, 1.73, 1.66, 这也意味着在锗晶体中材料的塑性随着锑空位复合体的引入得到提高。 泊松比是描述了材料的横向变形的弹性弹性常数, 常用来表征晶体在剪切应力下的稳定性, 由以下公式求得
[20 ]
:
ν = (3B -2G ) / (6B +2G ) (11)
泊松比值越小, 意味着晶体在剪应力下更好的稳定性, 随着锑空位缺陷的引入, 经过计算发现泊松比在0.24~0.25范围内, 比较纯锗的0.21, 有微弱的提升。 Pettifor
[26 ]
提出晶体中原子间的键角特征会影响材料的延性与脆性, 可以使用柯西压力 (C 12 -C 44 ) 来描述, 若柯西压力数值大于0, 可认为材料为韧性, 反之则材料呈现脆性
[25 ]
, 且随着柯西压力绝对值的增加, 材料的韧性/脆性增大。 无缺陷锗晶体的柯西压力值为-18.98 GPa, 具有本征脆性, 而随着锑空位复合体的引入, 柯西压力值逐渐增加如表5所示, 这也意味其塑性的加强。
3 结 论
1. 对于单空位缺陷锑掺杂锗晶体而言, 空位倾向于扩散到锑原子的第一近邻位, 进而形成锑空位复合体。
2. 当体系中含有两个空位时, 空位之间相互吸引, 并扩散到锑原子附近即空位对处于锑同一共价键方向的的第一、 二近邻晶格位点。 在此情形模型最稳定, 这也与晶体学中具有负体积空位倾向于偏聚进而减小基体界面能的结论相一致。
3. 与无缺陷锗晶体的柯西压力值为-18.98 GPa相比, 随着锑空位复合体的引入, 不同的结构模型的柯西压力始终为负且不断增大, 这也意味着锗晶体材料塑性的增大。
4. 依照Pugh判据分析, 无缺陷锗晶体的B /G 值为1.38, 随着锑空位复合体的引入B /G 值逐渐上升到1.59~1.73范围之内, 但并未达到临界值1.75。 说明由于锑空位复合体的引入使得锗晶体材料的脆性得到改善, 塑性得到提高, 对于锑掺杂锗晶体的实际加工过程具有重要指导意义。
参考文献
[1] Impellizzeri G, Boninelli S, Priolo F, Napolitani E.Fluorine effect on As diffusion in Ge [J].Journal of Applied Physics, 2011, 109 (11) :599.
[2] Markevich V P, Peaker A R, Hamilton B, Litvinov V V, Pokotilo Y M, Lastovskii S B, Coutinho J, Carvalho A, Rayson M J, Briddon P R.Tin-vacancy complex in germanium [J].Journal of Applied Physics, 2011, 109 (8) :201.
[3] Chroneos A, Bracht H, Grimes R W, Uberuaga B P.Vacancy-mediated dopant diffusion activation enthalpies for germanium [J].Applied Physics Letters, 2008, 92 (17) :172103.
[4] Chroneos A, Grimes R W, Tsamis C.Atomic scale simulations of donor-vacancy pairs in germanium [J].Journal of Materials Science Materials in Electronics, 2007, 18 (7) :763.
[5] Markevich V P, Hawkins I D, Peaker A R, Emtsev K V, Emtsev V V, Litvinov V V, Murin L I, Dobaczewski L.Vacancy-group-V-impurity atom pairs in Ge crystals doped with P, As, Sb, and Bi [J].Physical Review B, 2004, 70 (70) :155.
[6] Chroneos A, Grimes R W, Uberuaga B P, Bracht H.Diffusion and defect reactions between donors, C, and vacancies in Ge.II.Atomistic calculations of related complexes [J].Physical Review B, 2008, 77 (23) :235208.
[7] Brotzmann S, Bracht H.Intrinsic and extrinsic diffusion of phosphorus, arsenic, and antimony in germanium [J].Journal of Applied Physics, 2008, 103 (3) :3275.
[8] Tahini H, Chroneos A, Grimes R W, Schwingenschl?gl U, Bracht H.Diffusion of E centers in germanium predicted using GGA+U approach [J].Applied Physics Letters, 2011, 99 (7) :33508.
[9] Chen J, Wu T, Ma X, Wang L, Yang D.Ge-vacancy pair in Ge-doped Czochralski silicon [J].Journal of Applied Physics, 2008, 103 (12) :123519.
[10] Wang J, Fang F, Zhang X.An experimental study of cutting performance on monocrystalline germanium after ion implantation [J].Precision Engineering, 2015, 39:220.
[11] Fujita F E.Generation of vacancies in high-speed plastic deformation [J].Materials Science & Engineering A, 2003, 350 (2) :216.
[12] Zhu P P, Guo X F, Cui H B.Effect of point defects on physical and mechanical properties of B2-CoSc intermetallic studied by first-principles method [J].The Chinese Journal of Nonferrous Metals, 2017, (8) :1589. (朱攀攀, 郭学锋, 崔红保.点缺陷对B2-CoSc化合物物性影响的第一性原理研究 [J].中国有色金属学报, 2017, (8) :1589.)
[13] Clark S J, Segall M D, Pickard C J, Hasnip P J, Probert M I J, Refson K, Payne M C.First principles methods using CASTEP [J].Zeitschrift für Kristallographie, 2005, 220 (5) :567.
[14] Pfrommer B G, C?té M, Louie S G, Cohen M L.Relaxation of crystals with the quasi-newton method [J].Journal of Computational Physics, 1997, 131 (1) :233.
[15] Singh H P.Determination of thermal expansion of germanium, rhodium and iridium by X-rays [J].Acta Crystallographica, 1968, 24 (4) :469.
[16] Mattsson T R, Mattsson A E.Calculating the vacancy formation energy in metals:Pt, Pd, and Mo [J].Physical Review B Condensed Matter, 2002, 66 (21) :214110.
[17] Chroneos A, Uberuaga B P, Grimes R W.Carbon, dopant, and vacancy interactions in germanium [J].Journal of Applied Physics, 2007, 102 (8) :473.
[18] Chroneos A.Isovalent impurity-vacancy complexes in germanium [J].Physica Status Solidi., 2007, 244 (9) :3206.
[19] Chroneos A, Grimes R W, Uberuaga B P, Brotzmann S, Bracht H.Vacancy-arsenic clusters in germanium [J].Applied Physics Letters, 2007, 91 (19) :195203.
[20] Nichols C S, Van de Walle C G, Pantelides S T.Mechanisms of dopant impurity diffusion in silicon [J].Physical Review B, 1989, 40 (8) :5484.
[21] Hill R.The elastic behaviour of a crystalline aggregate [J].Proceedings of the Physical Society, 2002, 65 (5) :349.
[22] Zhao H, Zhao Y H, Yang X M, Sui H M, Hou H, Han P D.First-principles study on elastic properties and electronic structure of Ca, Sr and Ba doped Mg2 Si [J].Rare Metal Materials and Engineering, 2015, (3) :638. (赵慧, 赵宇宏, 杨晓敏, 眭怀明, 侯华, 韩培德.Ca, Sr和Ba掺杂Mg2 Si弹性性能和电子结构的第一性原理研究 [J].稀有金属材料与工程, 2015, (3) :638.)
[23] Fulcher B D, Cui X Y, Delley B, Stampfl C.Hardness analysis of cubic metal mononitrides from first principles [J].Physical Review B, 2012, 85 (18) :1614.
[24] Chen S, Lu J S, Xie M, Xia L, Pan Y, Hu J Q.First-principle investigation on mechanical performances of platinum-rhodium alloys [J].Chinese Jounal of Rare Metals, 2015, 39 (3) :276. (陈松, 陆建生, 谢明, 夏璐, 潘勇, 胡洁琼.铂铑合金系基本力学性能的第一性原理研究 [J].稀有金属.2015, 39 (3) :276.)
[25] Pugh S F.XCII.Relations between the elastic moduli and the plastic properties of polycrystalline pure metals [J].Philosophical Magazine, 2009, 45 (367) :823.
[26] Pettifor D G.Theoretical predictions of structure and related properties of intermetallics [J].Metal Science Journal, 1992, 8 (4) :345.