文章编号:1004-0609(2008)03-0516-07
Aln+(n=2~13)团簇分裂机理的计算
李贵发,彭 平,周惦武,韩绍昌
(湖南大学 材料科学与工程学院,长沙 410082)
摘 要:基于Aln+m+团簇分离方式主要以离解出中型或带电单Al原子为主的实验结果,采用线性同步转变(LST)和二次同步转变(QST)方法考察了Aln+m+ (n+m≤13) 团簇在双分模式下的不同分裂过程,并通过分裂吸收热?HR-P和分裂激活能?ER-T等的计算,在能态结构上分析了Aln+(n=2~13)团簇的分裂路径及其机理。结果表明: Aln+ (n=2~13)团簇以分裂出电中性或带电Al原子模式吸热?HR-P和所需分裂激活能?ER-T最少,而分裂成原子数相差较小的两个较大团簇所需裂解能量最多,因此,Aln+ (n=2~13)团簇主要以Aln+ →Al+ Aln-1+或Aln+→Al++ Aln-1路经裂解。
关键词:Aln+团簇;分裂机理;密度泛函理论;线性同步转变方法
中图分类号:O 641 文献标识码:A
Calculation of decomposition mechanism of Aln+(n=2~13) clusters
LI Gui-fa, PENG Ping, ZHOU Dian-wu, HAN Shao-chang
(School of Materials Science and Engineering, Hunan University, Changsha 410082, China)
Abstract: Based on the experimental result of Aln+m+ clusters decomposed by means of a isolated Al atom or cation, the disassociation route and mechanism of Aln+m+ (n+m≤13) clusters in the Aln ++Alm (n=1-12, m=1-12) mode were investigated by linear synchronous transit(LST)and quadratic synchronous transit (QST) method. Several parameters, such as the ionization potential, the endothermic reaction heat ?HR-P and the dissociation barrier energy ?ER-T of Aln+m+ (n+m≤13) clusters were calculated. Comparison of ?HR-P and ?ER-T requested in the disassociation route reveals the least energy of a isolated Al atom or cation from the Aln+m+ clusters is related to bigger ?HR-P and ?ER-T values, while a big cluster decomposites into two small clusters. The energetics difference between routes should be responsible for the preferential route of dissociation of Aln+ (n=2-13) clusters in terms of Aln+ →Al+ Aln-1+ or Aln+→Al++ Aln-1.
Key words: Aln+ cluster; decomposition mechanism; density functional theory; linear synchronous transit
团簇作为特殊物相,近年引起了人们广泛的研究兴趣。采用气体冷凝、磁控溅射、激光热解及溶胶凝胶等方法,人们已制备出多种电中性或带电的金属团簇[1],并通过对其离化能[2]与结合能[3]等结构参数的测试与计算,考察了其稳定的结构形态[4]、裂解方式[1]以及原子数n对其电磁特性的影响[5-6]等。对于Aln+团簇的裂解,也有很多学者对此进行过研究,如JARROLD等[7]在惰性气体氩环境中以5.25 eV的轰击能量采用撞击分离(Collision induced dissociation, CID)方法研究了Aln+ (n=3~26)团簇的离化能和分离方式,HANLEY等[8] 和RAY等[3]在惰性气体氙气氛下、分别以0~10 eV和1.88~6.99 eV轰击能量研究了Aln+(n=2~7)和Aln+ (n=7~17)团簇的裂解特性。虽然不同实验测得的分离能绝对值不等,但总体趋势一致,即Aln+团簇的分离方式主要以分离中型或带电单Al原子为主。后来,ING?LFSSON等[9]进一步在惰性气体氩气氛下,以0.1~10 eV为轰击能量,用CID方法研究了Aln+(n=2~11)团簇的分裂行为,并采用分子轨道理论对Al8+团簇的分离行为进行了分析,确认从Aln+(n=2~11)团簇中分离出中型或带电单Al原子为其主要模式。为了分析和更好地理解这些团簇主要以这种方式进行分解,本文作者采用基于密度泛函的分子轨道理论,通过化学反应动力学的方法,计算了在两相分离模式下Aln+(n=2~13)团簇不同分裂路径时的分裂吸收热与分裂激活能,以揭示Aln+(n=2~13)团簇的分离机制和深入开展对Aln+团簇分解演化行为的研究。
1 计算模型与方法
基于RAO 等[4]和LLOYD等[10]的研究结果,构造了如图1所示的稳态Aln(n=2~13)团簇计算模型,初始Al―Al键长设为d=0.286 3 nm。计算采用基于密度泛函理论(Density function theory, DFT)的Dmol程序。几何优化与总能计算时,电子交换关联能函数采用GGA近似的PBE形式[11],势函数取全电子位势,电子波函数采用带d轨道的双数值基(Double-numerical quality basis set, DN) 函数[12],布里渊区积分采用Monkhorst-Pack形式的特殊K点方法[13]。自洽(Self-consistent field, SCF)计算时,体系总能量和电荷密度收敛精度设为10-5原子单位。能量计算前先进行几何优化,以取得团簇模型的局域稳定结构,优化时其精度设置为:能量不大于50 μeV/atom,应力不大于1 eV/nm,位移不大于0.02 nm。计算时对团簇的对称性进行限制。然后,采用线性同步转变(Linear synchronous transit, LST)与二次同步转变(Quadratic synchronous transit, QST)方法[14]进行分裂模拟和过渡态结构搜索。为了简化,本文作者仅考察了由一个大团簇分裂为两个小团簇的“双分模式”。
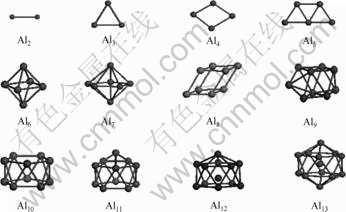
图1 Aln+(n=2~13)团簇计算模型
Fig.1 Calculation models of Aln+(n=2~13) clusters
在分裂模拟之前,首先选取Al2团簇测试团簇间的有效分离距离,测试结果如图2所示。由图2可见,当两个团簇间的中心距离D不小于0.7 nm时,体系总能量趋于恒定,即可认为是两个无相互作用的独立团簇,为此,本文作者在采用LST/QST进行团簇生长模拟时,0.7 nm被设置成了独立团簇间的最低间距。
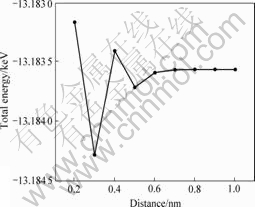
图2 双Al2团簇体系的总能量与两团簇中心间距的关系
Fig.2 Relationship between total energies and distance of two Al2 clusters
线性同步转变与二次同步转变方法是HALGREN等[14]于1977年提出的研究化学反应路径的方法,其计算方法如图3所示。首先给出团簇反应物-生成物的分离路径,在此路径上取一定数量的化学反应点2~5,连接反应物1点和生成物6点,此即LST路径。然后,在LST路径上找到能量最大的点4,应力是能量对位移的偏导数,而过渡态结构对应 “山脊”的力为零,即能量对位移的偏导数为零时,又可以找到一条力(F)为零的路线4-5′-3″,直到找到一个比LST路径中高能量点4要小的第二个高能量点3″,这条计算路径被称作共轭梯度搜索。最后再以3″为最高的能量 点,按照线性同步转变(LST)计算方法进行计算1-2″-3″-6各点对应团簇结构的能量,即二次同步转变(QST)。循环搜索,直到找到过渡态结构对应的能量点为止[14]。

图3 LST/QST计算方法示意图
Fig.3 Schematic diagram of LST/QST method
2 分析与讨论
2.1 Aln+ (n=2~13)团簇离化势
作为测试,本文作者采用下式计算了Aln+(2~13)团簇的离化势(EIP) [4]:

计算结果如图4所示。

图4 Aln+(2~13) 团簇离化势与计算值[4]和实验值[2]的比较
Fig.4 Comparison of ionization potentials of Aln+(2-13) clusters with experimental data[2] and calculated values[4]
由图4可见,虽然本研究计算的离化势(EIP)比实验值[2]及RAO等[4]采用Gaussian程序GGA近似得到的结果略低,但误差均在5%的范围内。在n为4,6, 8和13时,EIP出现峰值;在n为7和10时,EIP出现谷值,主要是源于Aln团簇电子壳层结构的幻数特性,即Aln团簇在n为6和13时,外层电子为18和39,难失去一个电子而变成带正电团簇,因此,离化势较高;而当n=7时,外层电子为21,易失去一个电子而成为满壳层结构,因而离化势较低;当n为4,8和10时,离化势不能很好地用外层电子数解释,还要考虑到团簇几何结构的影响。三者在整体变化趋势上一致表明本研究采用的计算方法与条件基本合适。
2.2 Aln团簇的分裂
Aln团簇分裂有多种模式,除稳定组态外,Aln (n=2~13)团簇如Al6还存在某些亚稳结构[15],作为团簇分裂机制研究的第一步,本研究在此仅考察了稳态Aln+ (n=2~13)+ 团簇的双分模式,即Aln+m + ?Aln+ + Alm模式,其中Aln+m +为团簇分裂反应物;Aln+和Alm为团簇分裂生成物;n取为1~12;m取为1~12,n+m≤13。采用线性同步转变(LST)与二次同步转变(QST)方法计算得到的这种双分模式下体系总能量随路径参数的变化曲线如图5所示。由图5可见,Aln+m+分裂成Aln+和Alm时体系总能量升高,表明Aln+ (n=2~13)团簇双分模式下的分裂为一吸热过程;在部分分裂模式下,团簇分裂出现了能垒,表明其间存在过渡态。根据化学反应动力学可知,反应吸收热和反应活化能是表征化学反应进行难易程度的两个重要参数,即反应吸收热越多与反应活化能越大,则需要外界给予的能量越多,反应就相对难以进行。为此,本文作者采用下式计算了这种双分模式下的分裂吸收热?HR-P与分裂激活能?ER-T:

计算结果如表1所列。
表1 Aln+m+ (n=1~12, m=1~12)团簇在Aln++Alm分裂模式下的反应吸收热?HR-P 与分裂激活能?ER-T
Table 1 Endothermic reaction energies ?HR-P and dissociation activation energies ?ER-T of Aln+m+ (n=1-12, m=1-12) clusters in Aln++Alm mode (eV/atom)
`
比较表1中本研究计算值与ING?LFSSON等[9]采用CID方法测量的实验值可见:本研究计算值比实验值稍小,因第一原理计算的是基态团簇的能量,忽略了实验中温度的影响。对于Alm +→Al + +Alm-1分离模式,当m=2时,计算值与实验值差别较大;当m为3~10时,则基本相同;从m=5开始,吸收热?HR-P呈递减趋势;对于Alm+ →Al2+Alm-2+ 和Alm+→Al3+Alm-3+分离模式,实验值比计算值稍大,但都在Al7+处出现了极大值,两边呈递减趋势。由于Al7+团簇外层电子数为20,满足闭壳层结构而表现出高稳定性,较难分裂。总体上看,Aln+m+团簇以分离中性或带单位电荷的Al原子为主,与实验结果[9]变化趋势基本一致。
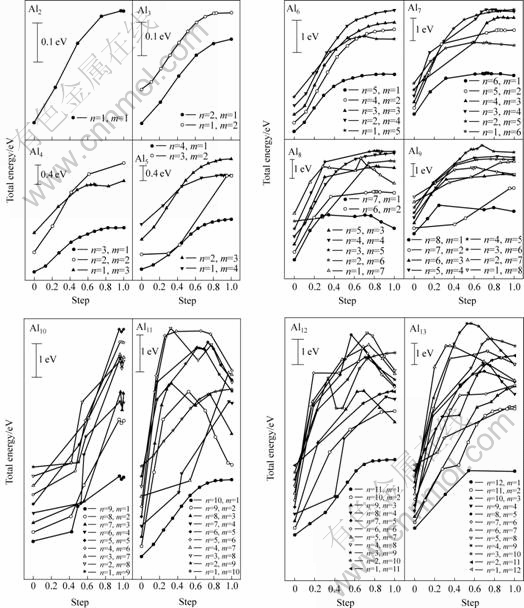
图5 Aln+m + (n=1~12, m=1~12) 团簇在Aln ++Alm 分裂模式下体系能量随分裂轨迹变化示意图
Fig.5 Schematic diagrams of total energies of system with path coordinates in bi-dissociation mode Aln+m+ ? Aln++Alm
本研究将基于图5与表1的计算结果,进一步对Aln+(n=2~13)团簇的分裂模式进行分析。
2.2.1 Al2+~Al5+团簇
对于Al2+团簇,只有一种分裂模式,即分离出一个中性和一个带正电的Al原子。对于Al3+团簇,分裂有两种情况,即分离出一个中性Al原子或分离出一个带单位电荷的Al原子。由图5(a)可见,它们都是不需克服能垒的吸热反应,比较其分裂吸收热(?HR-P)(见表1)可见:Al2+团簇的分离吸收热?HR-P=0.141 eV/atom,比Al3+团簇分离吸收热?HR-P(0.312 eV/atom和0.341 eV/atom)小,说明Al2+是一个活跃性较强的团簇,因此,实验中检测到的Al2+团簇比Al3+团簇多[9]。对于Al4+与Al5+团簇,其分离除了产生一个中性或带单位电荷的Al原子这种简单模式外,还出现了一种分离成两个小团簇的模式,即分离成Al2+和Al3+。由图5(a)可见,不论是分裂成原子的简单模式,还是分裂成大团簇的复杂模式,它们都不需克服能垒,比较表1中其分离反应热?HR-P可见,Al4+?Al2+ +Al2比Al4+?Al++Al3和Al4+?Al+Al3+ 吸热多,Al5+?Al2++ Al3与Al5+?Al2+Al3+比Al5+?Al++Al4和Al4+?Al+ Al4+吸热多,表明小团簇分裂出带电与电中性单Al原子比分裂成两团簇要相对容易。
2.2.2 Al6+ ~ Al7+团簇
由图5(b)可见,与面型Al2~Al4团簇类似,体型Al6团簇的分裂也是不需克服能垒的吸热过程,但同为体型结构的Al7团簇,其分裂则与Al2~Al6团簇不同,在分裂过程中出现了过渡态,分裂过程需克服一定能垒。对于Al6团簇,共有5种分裂方式:分别分裂成Aln+ (n为1, 2, 3, 4, 5)。由表1可见,分裂吸收热?HR-P最小的是分裂出中性或带单位电荷Al原子的模式,即Al6+?Al++Al5和Al6+?Al+Al5+,?HR-P分别为0.299与0.301 eV/atom。但比较两者可知,分离出Al+又相对容易些。对于Al7+团簇的6种可能分裂方式,大部分分裂路径存在反应能垒?ER-T,而有部分分裂路径(如Al7+?Al2+ +Al5和Al7+? Al5+ +Al2)则只有分裂吸收热?HR-P。由于能垒只是使分裂需要的热能有所增加,即额外增加(?ER-T-?HR-P)热能,因此,对于存在能垒的团簇分解,比较其反应能力只需用其反应能垒?ER-T替代其分裂吸收热?HR-P,如Al7+?Al+ +Al6和Al7+?Al+Al6+ 模式,其分裂吸收热?HR-P分别为0.246与0.247 eV/atom,但由于分别存在分解能垒?ER-T= 0.247与0.262 eV/atom,?ER-T分别比?HR-P高3与15 meV/atom,因此其反应能力的比较将只比较其?ER-T的大小。从表1可看出,Al7+团簇分裂需要外界提供能量最少的分解路径是Al7+?Al++Al6和Al7+?Al+ Al6+ 模式,即分裂出中性或带单位电荷Al原子的模式,所需能量分别为0.247和0.262 eV/atom;分离出Al+比分离出Al相对容易,其结果与面型Al2+~Al5+团簇类似。
比较图1所示Al6+与Al7+团簇的几何结构可见,Al6+ 团簇为八面体结构,而Al7+团簇是十面体结构。前者具有晶体结构单元特性,后者具有非晶与准晶结构单元特征。研究结果表明:无论是具有晶体结构特性的团簇,还是具有非晶或准晶结构特征的团簇,其带正电团簇的分解都趋向于分裂出中性或带单位电荷Al原子为主。
2.2.3 Al8+ ~ Al9+团簇
与八面体结构Al6+团簇一样,具有C2h对称性的Al8+团簇与具有C2v对称性的Al9+团簇,图5(b)显示其团簇分裂大部分为需要克服能垒的吸热反应。对于Al8+团簇,除分裂成Al4++Al4外,其他分裂方式都存在分裂能垒。比较表1中分裂反应热?HR-P和反应能垒?ER-T发现,分裂成大团簇的Al4++Al4模式需要能量最多,达0.547 eV/atom,而分裂出中性和带单位电荷Al原子时所需能量最少,分别为0.294与0.364 eV/atom。对于Al9+团簇,除分裂成Al2++Al7和Al2+Al7+外,其他分裂方式都存在分裂能垒,其中需能最多也是分裂成大团簇Al5++Al4或Al4++Al5的分解路径,分别为0.530与0.586 eV/atom,需能最少也是分裂出中性或带电荷Al原子的分解路径,分别为0.243与0.256 eV/atom。与Al2+~Al6+团簇不同,最可能的分解路径不是分离出带电Al+原子,而是分离出电中性Al原子,这与ING?LFSSON等[9]实验测试结果一致。
2.2.4 Al10+~ Al13+团簇
由图1可见,Al10+~Al13+团簇都具有某些5重对称性元素,因此,也是非晶性团簇。由图5(c)可看出,Al10+团簇的分解除Al5++Al5 路经外,其他路径都出现了过渡态,需能最多的分解路径为Al5++Al5,?HR-P=0.510 eV/atom,需能最少的分解路径为Al++Al9与Al+Al9+,?ER-T分别为0.190与0.191 eV/atom;而Al11+团簇除分裂成带电或电中性Al外,其他路径都存在分解能垒,前者也是需能最少的分解路经,?HR-P分别为0.188与0.190 eV/atom,而其中需能最多的分解路经为Al5++Al6与Al5+Al6+,?ER-T分别为0.432与0.429 eV/atom。
对于Al12+与Al13+团簇,图5(d)也显示了与上述Al11+团簇类似的形成特点,如分裂成结构相似的Al10+、Al11+和Al12+团簇时都没有出现能垒。分裂成两个较大的团簇时需能较多,分裂出带电或电中性单Al原子时需能最少,如Al12+团簇,需能最多的分解路经为Al5+Al7+,?ER-T为0.564 eV/atom,需能最少的分解路经为Al++Al11与Al+Al11+,?HR-P分别为0.246与0.241 eV/atom;Al13+团簇需能最多的分解路经为Al9+Al4+,?ER-T为0.581 eV/atom,需能最少的分解路经Al++Al11与Al+Al11+的?HR-P分别为0.208与0.214 eV/atom。
由此可见,非晶性团簇的分解,也趋向于分解成带电或电中性Al原子,裂解成两个较大的团簇则相对较难,但带电与电中性的Al原子裂出的优先性则不 明显。
3 结论
1) 对于Aln+(n=2~13) 团簇,双相分裂模式下的分裂吸收热?HR-P与分解激活能?ER-T 表明其主要以分裂成电中性或带电Al原子的裂解模式进行。
2) Aln+(n=2~13)团簇分裂成两个原子数相差不大的大团簇时所需能量最多,与其团簇合成相反,这种分裂模式不容易发生。
REFERENCES
[1] SAUNDERS W A, FAYET P, WOSTE L. Photodestruction of positively and negatively charged aluminum-cluster ions[J]. Phy Rev A, 1989, 39(9): 4400-4405.
[2] SCHRIVER K E, PERSSON J L, HONEA E C, WHETTEN R L. Electronic shell of group-III A metal atomic cluster[J]. Phys Rev Lett, 1990, 64(21): 2539-2543.
[3] RAY U, JARROLD M F, BOWER J E, KRAUS J E. Photodissociation kinetics of aluminum cluster ions: determination of cluster dissociation energies[J]. J Chem Phys, 1989, 91(5): 2912-2921.
[4] RAO B K, JENA P. Evolution of the electronic structure and properties of neutral and charged aluminum clusters: A comprehensive analysis[J]. J Chem Phys, 1999, 111(5): 1890-1904.
[5] HEER W A, MILANI P, CHATELAIN A. Nonjelliu-to-jellium transition in Aluminum cluster polarizabilities[J]. Phys Rev Lett, 1989, 63(26): 2834-2836.
[6] COX D M, TREVOR R L, WHETTEN R L. Aluminum clusters: Magnetic properties[J]. J Chem Phys, 1986, 84(8): 4651-4656.
[7] JARROLD M D, BOWER J E, KRAUS J S. Collision induced dissociation of metal cluster ions: Bare aluminum clusters Aln+(n=3-26)[J]. J Chem Phys, 1987, 86(7): 3876-3885.
[8] HANLEY L, RUATTA S A, ANDERSON S L. Collision- induced dissociation of aluminum cluster ions: Fragmentation patterns, bond energies, and structures for Al2+-Al7+[J]. J Chem Phys, 1987, 86(1): 260-268.
[9] ING?LFSSON O, TAKEO H. Energy-resolved collision- induced dissociation of Aln+ cluster (n=2-11) in the center of mass energy range from few hundred meV to 10 eV[J]. J Chem Phys, 1999, 110(9): 4382-4393.
[10] LLOYD L D, JOHNSTON R L. Modelling aluminium clusters with an empirical many- body potential[J]. Chem Phys, 1998, 236(1/3): 107-121.
[11] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18/28): 3865-3868.
[12] DELLEY B. Analytic energy derivatives in the numerical local- density-functional approach[J]. J Chem. Phys, 1991, 94(11): 7245-7250.
[13] PACK J D, MONKHORST H J. Special points for Brillouin- zone integrations―A reply[J]. Phys Rev B, 1977, 16(4/15): 1748-1749.
[14] HALGREN T A, LIPSCOMB W N. The synchronous-transit method for determining reaction pathways and locating molecular transition states[J]. Chem Phys Lett, 1977, 49(2): 225-232.
[15] 彭 平, 李贵发, 郑采星, 韩绍昌, 刘让苏. Aln(n=3, 4, 6, 13, 19)团簇的结构稳定性与形态演化[J]. 中国科学 E, 2006, 36(9): 975-982.
PENG Ping, LI Gui-fa, ZHENG Cai-xing, HAN Shao-chang, LIU Rang-su. The structure stability and configuration evolution of Aln(n=3, 4, 6, 13, 19) clusters[J]. Science in China Series E, 2006, 36(9): 975-982.
基金项目:教育部科技重点资助项目(104139);教育部博士点基金资助项目(20050532006)
收稿日期:2007-06-18;修订日期:2007-12-25
通讯作者:彭 平,教授,博士;电话:0731-8821610;E-mail: ppeng@hnu.cn
(编辑 李艳红)

