


Trans. Nonferrous Met. Soc. China 22(2012) 1190-1196
Characteristic atom sequences of Nb-Mo alloys system in BCC structure and properties of disordered alloys
PENG Hong-jian1,2, WU Qing1, LI Xiao-bo2, XIE You-qing2
1. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
2. School of Materials Science and Engineering, Central South University, Changsha 410083, China
Received 16 March 2011; accepted 20 February 2012
Abstract: Comprehensively considering energy, volume and electronic structure of alloys, the ninth equation was determined as the interaction function of Nb-Mo alloys system in BCC structure on the basis of idea of systematic science of alloys, experimental lattice constants and heats of formation of disordered Nb(1-x)Mox alloys. The structural parameters and properties of Nb and Mo characteristic atoms sequences and corresponding characteristic crystals sequences were determined in Nb-Mo alloys system. The electronic structure and physical properties of disordered Nb(1-x)Mox alloys system were calculated according to concentration of characteristic atoms of disordered alloys. The change trend of physical properties is the same as that of electronic structure.
Key words: Nb-Mo alloys system; characteristic atom sequences; disordered alloys; properties
1 Introduction
The target of material science development is to continuously design and develop new materials to meet modernization and future needs. To develop material design by multi-structure levels and various properties, materials science should be systematic. After nearly thirty years of efforts, XIE et al [1-12] had established the framework of systematic science of alloys on the basis of energy band theory, valence bond theory and statistical thermodynamics. The core is the theory of the characteristic crystals and points out direction of alloy design. The theory has been successfully applied to Ag-Cu alloys [1-4] and Au-Cu alloys [5-8] in FCC structure, Ti-Al alloys with coexistence of FCC and HCP structures [9-12].
However, there is little research on alloys in BCC structure by the theory. The purpose of the present work is to choose Nb-Mo alloys in BCC structure as research subject. Based on the experimental lattice constants, heats of formation of disordered Nb(1-x)Mox alloys and the theory of the characteristic crystals, comprehensively considering the energy, volume and electronic structure of alloys, the optimum equation was determined as the interaction function of Nb-Mo alloys system. The structural parameters and properties of Nb and Mo characteristic atoms sequences and corresponding characteristic crystals sequences in Nb-Mo alloys system were determined. According to the concentrations of the characteristic atoms, the structural parameters and properties of disordered Nb(1-x)Mox alloys were obtained. The framework of systematic science of alloys will be further developed and improved.
2 Models and laws
In the disordered solid solution, the concentrations of characteristic atoms are, respectively
 (1)
(1)
where 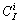 is the combination factor I!/[(I-i)!i!].
is the combination factor I!/[(I-i)!i!].
The average atomic energy of the alloys is
 (2)
(2)
The average atomic energies of components are, respectively
 (3)
(3)
 (4)
(4)
Substituting relations of property  of characteristic crystals of component A and i into Eq. (4), three relations of average property qA of component A with composition xA can be obtained (the derivation is omitted here):
of characteristic crystals of component A and i into Eq. (4), three relations of average property qA of component A with composition xA can be obtained (the derivation is omitted here):
For type I,
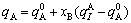 ; (5)
; (5)
For type II,
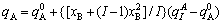 (6)
(6)
For type III,
 (7)
(7)
Similarly, three relations of average property qB of component B with composition xB can be obtained (the derivation is omitted here):
For type I,
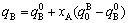 ; (8)
; (8)
For type II,
 (9)
(9)
For type III,
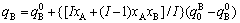 (10)
(10)
Combining the above equations and substituting them into Eq. (2), nine equations of average atomic potential energy and average atomic volume of solid solution can be obtained(see Table 1). For alloys system in BCC structure, I=8.
2.1 Determination of cohesive energies of Nb and Mo characteristic crystals of Nb-Mo alloys system in BCC structure
The crystalline structure of pure metals Nb and Mo is the same as that of Nb-Mo solid solutions.  and
and 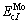 refer to cohesive energies of pure metals Nb and Mo, respectively.
refer to cohesive energies of pure metals Nb and Mo, respectively. and
and are calculated by interaction equations of binary disordered alloys [13] from heats of formation [14] of Nb(1-x)Mox solid solutions with different compositions. The results are listed in Table 2.
are calculated by interaction equations of binary disordered alloys [13] from heats of formation [14] of Nb(1-x)Mox solid solutions with different compositions. The results are listed in Table 2.
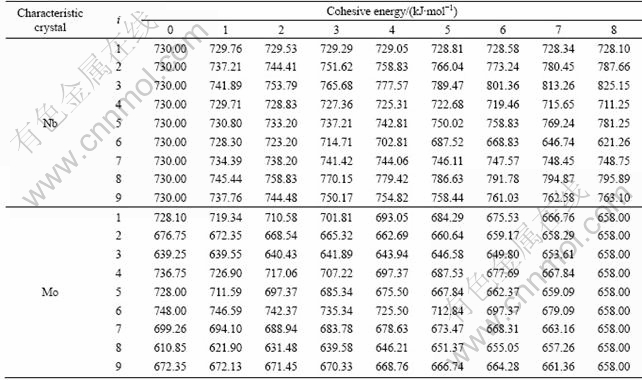
If we only consider the energy of disordered Nb(1-x)Mox alloy system in BCC structure, not average energy of every characteristic crystal, nine interaction equations are feasible in Table 1. The cohesive energies of the terminal state characteristic crystals are different from various equations. The cohesive energies of characteristic crystals by nine equations are listed in Table 3.
Table 1 Interaction equations of binary disordered alloys (q=E, v)

Table 2 Terminal and primary values of cohesive energies of Nb and Mo characteristic crystals of Nb-Mo alloys system
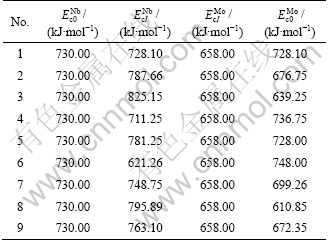
Table 3 Cohesive energies of characteristic crystals Nb and Mo of Nb-Mo alloys system by nine interaction equations
2.2 Determination of lattice constants of characteristic crystals Nb and Mo of Nb-Mo alloys system in BCC structure
The crystalline structure of pure metals Nb and Mo is also the same as that of Nb-Mo solid solutions.  and
and  refer to atomic volumes of pure metals Nb and Mo, respectively.
refer to atomic volumes of pure metals Nb and Mo, respectively. 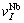 and
and 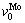 are calculated by volume interaction equations of binary disordered alloys from experimental lattice constants [15] of Nb(1-x)Mox solid solutions with different compositions. The results are listed in Table 4.
are calculated by volume interaction equations of binary disordered alloys from experimental lattice constants [15] of Nb(1-x)Mox solid solutions with different compositions. The results are listed in Table 4.
If we only consider the atomic volumes of disordered Nb(1-x)Mox alloys system in BCC structure, not average atomic volumes of every characteristic crystal, nine interaction equations are also feasible in Table 1. The atomic volumes of the terminal state characteristic crystals are different from various equations. The lattice constants of Nb and Mo characteristic crystals by nine equations are listed in Table 5.
The research on energy and volume interaction equations of alloys indicates that primary values are the same by different interaction equations. But cohesive energies of different characteristic crystals have great differences by different interaction equations. Therefore, every interaction equation describes a specific interaction essence of Nb and Mo atoms in the theory of characteristic crystals and reflects microscope essence of solid solution. It is incorrect that the interaction equations are chosen only from alloy energy as a function of composition. Only considering the relationship among energy, volume, atomic state of alloy, the correct interaction equation can be chosen. The ninth equation is determined as interaction equation of disordered Nb(1-x) Mox alloys by systematic analysis of properties of characteristic crystals in Tables 3 and 5.
 (11)
(11)
where =730 kJ/mol, =658 kJ/mol, = 763.10 kJ/mol, =672.35 kJ/mol, I=8.
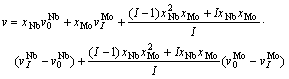 (12)
(12)
where =0.0179750 nm3,=0.0155893 nm3, =0.0175250 nm3, =0.0153085 nm3, and I=8.
Table 4 Terminal and primary values of volume of Nb and Mo characteristic crystals of Nb-Mo alloys system
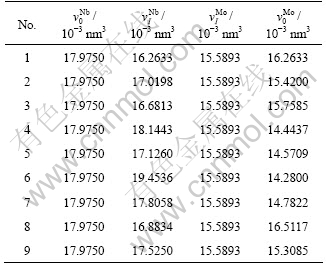
Table 5 Lattice constants of Nb and Mo characteristic crystals of Nb-Mo alloys system by nine interaction equations
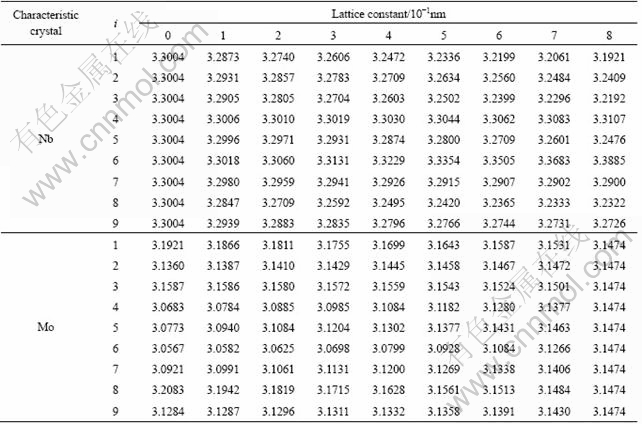
3 Nb and Mo characteristic atom sequences and characteristic crystal sequences of Nb-Mo alloys system in BCC structure
Structural parameters and properties of Nb and Mo characteristic atoms sequences and corresponding characteristic crystals sequences in Nb-Mo alloys system by experimental cohesive energies and lattice constants obtained by the ninth equation are listed in Tables 6 and 7. In the tables, dc, sc and pc are the number of covalent electrons in sЈ¬p, and d orbits; ec is the summation of covalent electrons; ef is the summation of free electrons; en is the summation of non-bonded electrons; R is the single radius; ri is the bond length of the ith neighbor bond; ni is the number of the ith neighbor bond; E1 is the bond energy in the nearest neighbor bond; Ec is cohesive energy.
4 Properties of disordered Nb(1-x)Mox alloys system in BCC structure
The properties of alloys are described by average properties of components. The properties of disordered alloys are obtained by additive law of characteristic crystals. Therefore, electronic structure and properties of disordered Nb(1-x)Mox alloys system as a function of composition xMo are listed in Table 8.
The electronic structures and properties of disordered Nb(1-x)Mox alloys system as function of composition xMo are shown in Figs. 1-3. As can be seen in Fig. 1, the number of covalent electrons ec increases and the number of free electron ef decreases with the concentration of Mo increasing. This is consistent with the cohesive energy. Figure 2 illustrates the relationship between bond length and composition. Single bond radius reduces with increasing the concentration of Mo. Figure 3 shows the relationship between the nearest neighbor bond length and bond energy. Bond length becomes short with increasing the concentration of Mo, electron pairs increase and bond energy decreases, and the change trend is very similar to that of electronic structure.
Table 6 Structural parameters and properties of Nb characteristic atom sequences and corresponding characteristic crystal sequences in Nb-Mo alloys system
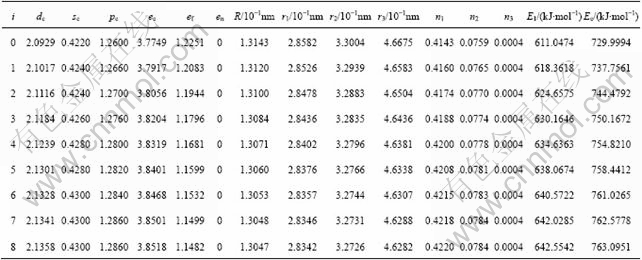
Table 7 Structural parameters and properties of Mo characteristic atom sequences and corresponding characteristic crystal sequences in Nb-Mo alloys system
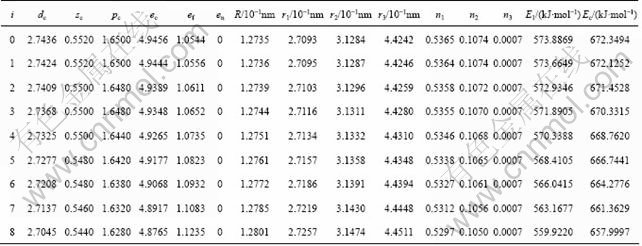
Table 8 Electronic structure and properties of disordered Nb(1-x)Mox alloys system as function of composition xMo
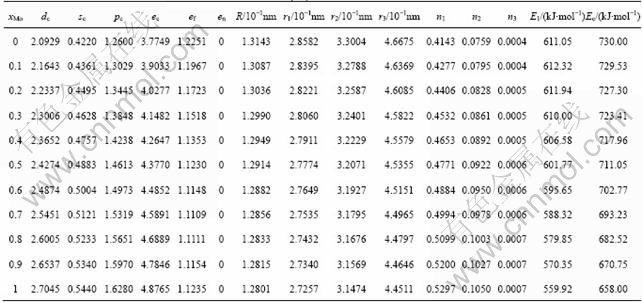
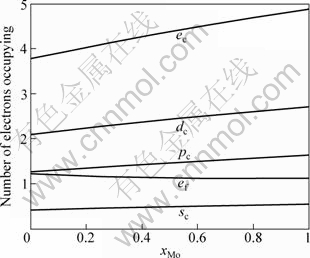
Fig. 1 Electronic structure of disordered Nb(1-x)Mox alloys system as function of composition xMo
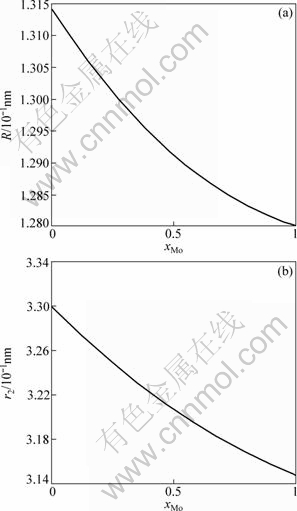
Fig. 2 Single bond radius (a) and lattice constants (b) of disordered Nb(1-x)Mox alloys system as function of composition xMo
5 Conclusions
1) On the basis of idea of systematic science of alloys and experimental lattice constants and heats of formation, comprehensively considering the energy, volume and electronic structure of alloys, the ninth equation was determined as the interaction equation of Nb-Mo alloys system.
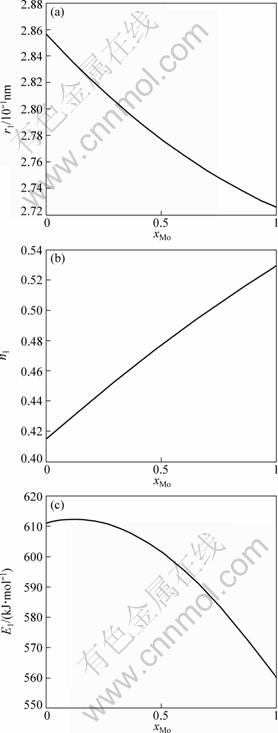
Fig. 3 The nearest neighbor bond length (a), number of electrons (b) and bond energies (c) of disordered Nb(1-x)Mox alloys system as function of composition xMo
2) The structural parameters and properties of Nb and Mo characteristic atoms sequences and corresponding characteristic crystals sequences were determined in Nb-Mo alloys system. The electronic structures and physical properties of disordered Nb(1-x)Mox alloys system were calculated according to concentration of characteristic atoms of disordered alloys and basic information of characteristic atom sequences and characteristic crystal sequences.
3) The change trend of physical properties is the same as that of electronic structure.
References
[1] XIE You-qing. Atomic energies and Gibbs energy functions of Ag-Cu alloys [J]. Science in China E, 1998, 41: 146-156.
[2] XIE You-qing, ZHANG Xiao-dong. Atomic volumes and volumes functions of Ag-Cu alloys [J]. Science in China E, 1998, 41: 157-168.
[3] XIE You-qing, ZHANG Xiao-dong. Electronic structure of Ag-Cu alloys [J]. Science in China E, 1998, 41: 225-236.
[4] XIE You-qing, ZHANG Xiao-dong. Phase diagram and thermodynamic properties of Ag-Cu alloys [J]. Science in China E, 1998, 41: 348-356.
[5] XIE You-qing, LIU Xin-bi, LI Xiao-bo, PENG Hong-jian, NIE Yao-zhuang. Volume sequences of characteristic atoms separated from experimental volumes of AuCu and AuCu3 compounds [J]. Transaction of Nonferrous Metals Society of China, 2009, 19: 1599-1617.
[6] XIE You-qing, LI Yan-fen, LIU Xin-bi, LI Xiao-bo, PENG Hong-jian, NIE Yao-zhuang. Systematic science of Au-Cu system based on experimental data of disordered alloys: Characteristic atom occupation patterns of Au3Cu, AuCu3, AuCu I and AuCu II [J]. Transaction of Nonferrous Metals Society of China, 2011, 21: 1092-1104.
[7] XIE You-qing, LI Xiao-bo, LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang. Volume sequences of characteristic atoms separated from experimental volumes of disordered Au(1-x)Cux alloys [J]. Journal of Materials Science and Engineering, 2009, 3: 54-75.
[8] XIE You-qing, LI Xiao-bo, LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang. Potential energy sequences of characteristic atoms on basis of heats of formation of disordered Au(1-x)Cux alloys [J]. Journal of Materials Science and Engineering, 2009, 6: 44-57.
[9] XIE You-qing, TAO Hui-jin, PENG Hong-jian, LI Xiao-bo, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC TiAl2 type alloys [J]. Physica B, 2005, 366: 17-37.
[10] XIE You-qing, PENG Hong-jian, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC Ti3Al-type alloys [J]. Physica B, 2005, 362: 1-17.
[11] XIE You-qing, LIU Xin-bi, PENG Kun, PENG Hong-jian. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC TiAl3-type alloys [J]. Physica B, 2004, 353: 15-33.
[12] XIE You-qing, PENG Kun, LIU Xin-bi. Influences of xTi/xAl on atomic states, lattice constants and potential energy planes of ordered FCC TiAl-type alloys [J]. Physica B, 2004, 344: 5-20.
[13] PENG Hong-jian, WU Qing, LI Xiao-bo, XIE You-qing, Computation of concentration of characteristic atoms of alloys in BCC structure [J]. Science in China E, 2011, 41: 1167-1174.
[14] SINGHAL S C, WORRELL W L. A high-temperature thermodynamic investigation of the Nb-Mo system [J]. Metall Trans, 1973, 4: 1125-1128.
[15] PEARSON W B. A handbook of lattice spacings and structures of metals and alloys [M]. London: Pergmon Press, 1958.
BCCЅб№№Nb-MoєПЅрПµµДМШХчФЧУРтБРј°ОЮРтєПЅрµДРФЦК
ЕнємЅЁ1,2Ј¬Од За1Ј¬АоРЎІЁ2Ј¬Р»УУЗд2
1. ЦРДПґуС§ »ЇС§»Ї№¤С§ФєЈ¬і¤Йі 410083Ј»
2. ЦРДПґуС§ ІДБПїЖС§У빤іМС§ФєЈ¬і¤Йі 410083
ХЄ ТЄЈєёщѕЭПµНієПЅрїЖС§µДЛјПлЈ¬ЅбєПКµСйѕ§ёсіЈКэєНЙъіЙИИЈ¬ЧЫєПїјВЗєПЅрДЬБїЎўМе»эЎўФЧУЧґМ¬Ц®јдµДП໥№ШПµЈ¬И·¶ЁBCC Ѕб№№Nb-MoєПЅрПµµДП໥ЧчУГ·ЅіМОЄµЪ9·ЅіМЈ»Н¬К±Ј¬И·¶ЁNb-MoєПЅрПµЦРNbєНMoМШХчФЧУРтБРєНМШХчѕ§МеРтБРµД»щ±ѕРЕПўЈ»ёщѕЭОЮРтєПЅрµДМШХчФЧУЕЁ¶ИјЖЛгБЛОЮРтNb(1-x)MoxєПЅрµДµзЧУЅб№№єНОпАнРФЦКЈ¬ЖдОпАнРФЦКµД±д»ЇЗчКЖУлµзЧУЅб№№µД±д»Їј«ЖдТ»ЦВЎЈ
№ШјьґКЈєNb-MoєПЅрПµЈ»МШХчФЧУРтБРЈ»ОЮРтєПЅрЈ»РФЦК
(Edited by YANG Hua)
Foundation item: Project (50954006) supported by the National Natural Science Foundation of China; Project (2009GK3152) supported by the Hunan Science and Technology Department, China; Project (201012) supported by the Hunan Provincial Construction Department, China; Project (K1003048-11) supported by the Changsha City Science and Technology Department, China
Corresponding author: PENG Hong-jian; Tel: +86-731-88879287; E-mail: phj108@163.com
DOI: 10.1016/S1003-6326(11)61304-6

