
Enhancement of photocatalytic activity of TiO2 film electrode by in situ photoelectro-generating active chlorine
CHENG Xiao-fang(程小芳)1, LENG Wen-hua(冷文华)1, PI Ou-yang(皮欧阳)1,
ZHANG Zhao(张 昭)1, ZHANG Jian-qing(张鉴清)1, 2, CAO Chu-nan(曹楚南)1, 2
1. Department of Chemistry, Zhejiang University, Hangzhou 310027, China;
2. State Key Laboratory for Corrosion and Protection of Metals, Institute of Metal Research,Chinese Academy of Sciences, Shenyang 110016, China
Received 31 December 2006; accepted 9 July 2007
______________________________________________________________
Abstract: The photoelectrocatalytic activity of TiO2 film electrodes in the degradation of nitrite ion was greatly enhanced in the presence of chlorine ion. The influences of NaCl concentration and initial pH value on the degradation rate of  and active chlorine production were studied. The results show that the decay rate of and the accumulation rate of active chlorine increase with increasing NaCl concentration. At pH<8, both the decay of and active chlorine formation rates are enhanced with increasing NaCl concentration, while at pH>10, they are suppressed. In addition, contrast to conventionally accepted view, in which an advantage of anatase over the rutile modification of TiO2 is in terms of photoactivity, it is found that a thermal oxidation rutile TiO2 electrode is more suitable for both photogenerating active chlorine and degrading in the presence of Cl-. The correlative mechanism was also discussed in detail. Specific adsorption of Cl- on the electrode causes its energy band edges to move towards positive value and also lower the photocurrent, thus less OH? radicals are produced. However, more active species of Cl? that have longer lifetime are available to take part in the oxidation of, thus improving its degradation rate.
and active chlorine production were studied. The results show that the decay rate of and the accumulation rate of active chlorine increase with increasing NaCl concentration. At pH<8, both the decay of and active chlorine formation rates are enhanced with increasing NaCl concentration, while at pH>10, they are suppressed. In addition, contrast to conventionally accepted view, in which an advantage of anatase over the rutile modification of TiO2 is in terms of photoactivity, it is found that a thermal oxidation rutile TiO2 electrode is more suitable for both photogenerating active chlorine and degrading in the presence of Cl-. The correlative mechanism was also discussed in detail. Specific adsorption of Cl- on the electrode causes its energy band edges to move towards positive value and also lower the photocurrent, thus less OH? radicals are produced. However, more active species of Cl? that have longer lifetime are available to take part in the oxidation of, thus improving its degradation rate.
Key words: TiO2; photoelectrocatalysis; active chlorine; nitrite ion
______________________________________________________________
1 Introduction
Photocatalysis is a promising advanced oxidation technology for the treatment of water. The photoelectrochemical(PEC) degradation of pollutants is an extension of heterogeneous photocatalytic progress, in which the catalyst is placed on an electrode that is controlled potentiostatically to make the photogenerated electron-hole pairs separate more efficiently[1-3]. Inorganic anions, such as Cl-,  and
and 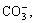 are commonly present in wastewater. Many researchers have investigated the effect of inorganic anions on the TiO2 photocatalytic degradation of pollutants[4-6]. Among them, Cl- was often studied[4-5]. Some researchers found that the presence of Cl- suppressed the degradation rate of organic pollutant, while others found a reverse result. For instance, LUO and HEPEL[4] found that addition of Cl- in acidic solutions could promote the photoelectrocatalytic degradation of naphthol blue black diazo dye. However, to the best of our knowledge, no systematic study has been carried out to clarify the role of Cl- in the process of photocatalytic reactions.
are commonly present in wastewater. Many researchers have investigated the effect of inorganic anions on the TiO2 photocatalytic degradation of pollutants[4-6]. Among them, Cl- was often studied[4-5]. Some researchers found that the presence of Cl- suppressed the degradation rate of organic pollutant, while others found a reverse result. For instance, LUO and HEPEL[4] found that addition of Cl- in acidic solutions could promote the photoelectrocatalytic degradation of naphthol blue black diazo dye. However, to the best of our knowledge, no systematic study has been carried out to clarify the role of Cl- in the process of photocatalytic reactions.
The electrolytic production of chlorine and hypochlorite from chloride is widely used in industry and for the disinfection of drinking water. Sometimes the electrochemical disinfection was based on the use of active chlorine produced as a main disinfecting agent from naturally occurring chloride ion[7-8]. Recently, it was found that it can be generated by a PEC method on a TiO2 thin film electrode under UV light illumination [9-10]. However, few studies[11] have monitored the in situ generated active chlorine during the PEC degradation of pollutants. And also no studies have been intentionally reported dealing with the possibility to use in situ photoelectrogenerated active chlorine to improve pollutants degradation rate.
It is well accepted that TiO2 is considered to be one of the most attractive photocatalysts for purifying and treating water, due to its excellent stability over a wide pH 0-14 and photoactivity under UV light. The most published work to date has dealt with the use of anatase form of TiO2 electrode[5,9,12]. The rutile TiO2 photoelectrode has seldom been investigated[13-14], which may be attributed to its low specific surface area, poor adsorption for oxygen and low photoactivity in photocatalytic reaction. Up to now, reports dealing with the application of rutile TiO2 in photoelectrocatalytically generating active chlorine are not found in the literature.
In this study, a thermal oxidation rutile TiO2 thin-film electrode was selected as the photoanode to degrade nitrite ion 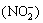 in aqueous solutions containing Cl-, and the concentration of active chlorine generated in situ was monitored. The most important parameters, such as types of TiO2, Cl- concentration and solution pH value, were investigated systematically. The relevant mechanism was discussed in detail.
in aqueous solutions containing Cl-, and the concentration of active chlorine generated in situ was monitored. The most important parameters, such as types of TiO2, Cl- concentration and solution pH value, were investigated systematically. The relevant mechanism was discussed in detail.
2 Experimental
2.1 Preparation of TiO2 thin-film photoelectrode
Two types of TiO2, i.e., thermal oxidation TiO2 (TO-TiO2) and sol-gel TiO2 (SG-TiO2) electrodes were prepared.
Titanium sheet (99.7%, 0.15 cm in thickness) was used as the substrate of photoanode, and its pre-treatment was the same as the previous reports[14]. The titanium sheets were annealed in a furnace in air to form the TO-TiO2 electrode as follows: increasing from room temperature to 600 ℃ at the rate of 5 ℃/min and then holding at this temperature for 1 h. As described in Ref.[13], a maximum photocurrent value under UV-light was attained at temperature of 600-700 ℃. The SG-TiO2 was fabricated by coating a layer of TiO2 sol onto the titanium substrates through repetitive heat treatment, i.e., the titanium substrates were dipped into TiO2 sol solutions for 30 s prepared by the method according to Ref.[13], pulled up and then heated for 10 min at 500 ℃ to coat the layer, repeated this procedure for 14 times and the resulting materials were fired at 500℃ for 1 h.
The annealed electrodes were electrically contacted with Cu wire on their backsides and covered with epoxy leaving a certain area for use. Photoelectrodes with working area of 20 cm2 (5 cm×4 cm) and 0.49 cm2 (0.7 cm×0.7 cm) were used for the degradation of  and photoelectrochemical measurements, respectively.
and photoelectrochemical measurements, respectively.
X-ray diffractometer analysis shows that the TO-TiO2 electrode sintered at 600 ℃ is in a rutile form of TiO2, while the SG-TiO2 electrode sintered at 500 ℃ has an anatase form[14].
2.2 Photodegradation of nitrite ions and photoelectro- generated active chlorine
Both the photodegradation of and the active chloride generated experiments were carried out in a two-compartment Teflon? cell equipped with a quartz window, which was described elsewhere[15]. A TiO2 electrode was used as the working electrode, with an area of 5 cm×4 cm exposed to the light, the counter electrode and the reference electrode were a large area specpure of graphite sheet and a saturated calomel electrode (SCE), respectively. The potential used in the text was specified to a SCE unless otherwise stated.
The photoanode was illuminated with a 300 W UV lamp (λmax=365 nm) filtered by quartz glass. The incident light intensity in the cell was estimated to be 6.5 mW/cm2 detected by an UV-irradiance meter (UV-A, Instruments of Beijing Normal University, China). All experiments were carried out on a ZF-5 Potentiosta/ Galvanostat (Shanghai Zhengfang Instrument, China).
In each experiment, 35 mL and 40 mL solutions were transferred into the anodic and cathodic compartments connected with a KNO3 salt bridge, respectively. Prior to illumination, all the solutions tested were stirred at least for 15 min by bubbling with air to obtain adsorption equilibrium, and kept purging during the experiments. Samples were periodically taken from the anodic compartment for analysis. The concentration of was analyzed spectrophotometrically at 540 nm[16]. The overall concentrations of dissolved chlorine in water, including three possible species: Cl2, HClO and ClO- here, were calculated in terms of active chlorine that was determined by the DPD (N, N-diethyl-p-phenylenediamine) colorimetric method [9-10].
The supporting electrolyte was 0.5 mol/L Na2SO4 with diluted H2SO4 and NaOH for pH adjustment, and NaCl was used as a source of Cl-. Unless otherwise specified, 10 mg/L was used.
was used.
2.3 Photocurrent measurements and Mott-Schottky (M-S) analysis
Both the photocurrent measurements and the M-S experiments were carried out in a three-electrode cell equipped with a quartz window without stirring. The TO-TiO2 was used as the working electrode, with an area of 0.7 cm×0.7 cm exposed to light, a platinum plate as the counter electrode, and a SCE as the reference electrode. In the photocurrent measurements, the excitation light source was a 500 W Xe lamp with a 10 cm×10 cm×20 cm Pyrex? glass vessel of water in front of the lamp to minimize heating. All experiments were carried out on CHI660 electrochemical station (Shanghai Chenhua Corp., China) driven by a personal computer. In the M-S analysis the light source was a 250 W xenon lamp (Müller Elecktronik Optik, Germany), and more detailed procedures can be obtained in Ref.[15].
The electrolyte was 0.5 mol/L Na2SO4 (pH 4.0) with a desired amount of NaCl added, and all other chemicals and solvents were of reagent grade and used without further purification. Doubly distilled water was used in all experiments.
3 Results and discussion
3.1 Blank test
In the blank tests it is found that under the open circuit conditions, i.e., without applying a potential, no degradation of was detected with the TO-TiO2 under UV light irradiation both in the solutions with and without Cl-. No removal of was found after electrolysis of the solution without illumination even if the applied potential was up to +1 V. No noticeable degradation of was detected in the presence of NaCl but without illumination at +1.0 V, which excluded the effect of electrochemically oxidized of chlorine ion. All of these observations indicate that the oxidation of results completely from the electrochemically assisted photocatalytic reaction.
3.2 Effect of Cl- concentration
The effect of chloride ions concentration on the removal of as a function of time is shown in Fig.1(a). It can be seen that the rate of degradation of increases rapidly with increasing concentration of Cl-. For instance, in the absence of NaCl, only 70% of is oxidized within 20 min, while adding 0.01 mol/L NaCl, complete degradation of is achieved within 15 min. When the concentration of NaCl reaches 0.5 mol/L, only 4 min is needed for complete degradation.
The photoelectrocatalytic production of active chlorine as a function of the concentration of NaCl was also investigated using 0.5 mol/L Na2SO4 (pH 4.7) solutions at +1.0 V. The amount of active chlorine produced was monitored in interval of 6 min during the entire 30 min. The results are shown in Fig.1(b). High active chlorine production is obtained with the increase of chloride ions concentration.
3.3 Effect of pH
The removal rates of at different initial pH values are plotted in Fig.2(a) as a function of time. As shown in solutions without Cl-, the conversion of nitrite decreases as follows: pH 10.5>6.2>4.7. A similar result was also reported by others[16]. However, in solutions with Cl-, this conversion increases with decreasing pH value.
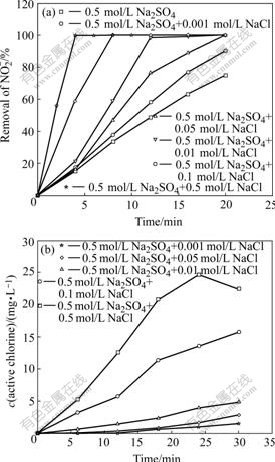
Fig.1 Photoelectrochemical degradation of  on TO-TiO2 electrode at +1.0 V in 0.5 mol/L Na2SO4 solution (pH 4.7) containing different NaCl concentration (a) and dependence of active chlorine generation rate on NaCl concentration on TO-TiO2 electrode at +1.0 V (b)
on TO-TiO2 electrode at +1.0 V in 0.5 mol/L Na2SO4 solution (pH 4.7) containing different NaCl concentration (a) and dependence of active chlorine generation rate on NaCl concentration on TO-TiO2 electrode at +1.0 V (b)
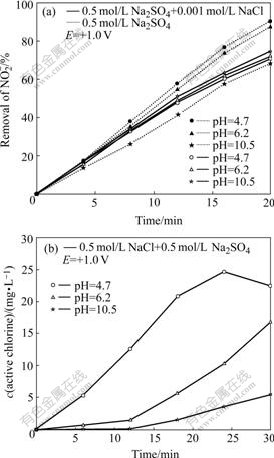
Fig.2 Effect of pH on PEC removal of on TO-TiO2 electrode (a) and dependence of active chlorine generation rate on pH on TO-TiO2 electrode (b)
The influence of initial pH on active chlorine production was also investigated. As shown in Fig.2(b), the rate is the highest at pH 4, and decreases sharply at pH 6.2, and completely decays when the pH value reaches 10.5. Similar results were obtained on an anatase TiO2 by other researchers, and the interpretation is that in basic media, the photogenerated holes are mainly involved in the hydroxyl radical formation, thus producing less active chlorine[9].
3.4 Mechanism of enhanced performance
Generally, heterogeneous photocatalytic degradation mechanism of pollutants can be classified into two types: the free radical mechanism (indirect) and the valence-band hole direct oxidation mechanism. The PEC degradation of was reported to proceed according to the indirect mechanism under UV light[16]. The mechanisms of PEC oxidation of nitrite ion by OH radicals can be proposed as
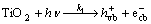 (1)
(1)
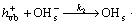 (2)
(2)
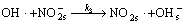 (3)
(3)
where the subscript ‘vb’ is the valence band, ‘cb’ is the conduction band and ‘s’ means adsorbtion on the surface.
It is found that there is no change in the rate upon further increasing the stirring rate of the solutions, indicating that the mass transport of is not the rate-determining step. By applying an anodic potential as high as +1.0V, the photogenerated electrons can be transferred through the external circuit efficiently, thus we can exclude the probability of cathodic reaction controlling. So the decay rate, R, can be simply expressed as
 (4)
(4)
There is no noticeable adsorption of on the electrode detected at pH 6.3, so its surface concentration is supposed to be equal to that in the bulk solution when rigorously stirring the solution in the experiments. Also, in most experiments, the concentration of is 10 mg/L, much larger than the flux of photons (and therefore [OH?]). Thus the R is mainly determined by the k3 and the concentration of OH?.
can also be degraded by the active chlorine photogenerated[7], and these processes can be denoted as
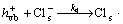 (5)
(5)
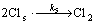 (6)
(6)
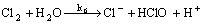 (7)
(7)
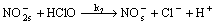 (8)
(8)
Then R at this case can be expressed as
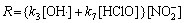 (9)
(9)
From Eqns.(4) and (9) we can not conclude that factor is responsible for the promotion of photodegradation rate of in the presence of Cl-. To clarify this, firstly, the photocurrents at +1.0 V in the solutions of pH 4.0 with different [Cl-] were measured and the results are shown in Fig.3. Obviously, the photocurrent decreases with addition of Cl-. Secondly, the M-S analysis was carried out to infer whether the energy band edges of the electrode were shifted with Cl- in the solutions, since its flat-band potential(Efb) can be calculated from the M-S equations[15]. As shown in Fig.4, Efb shifts slightly towards positive value with the increase of Cl-. This suggests the valence band edges of the electrode also move in the same direction, consequently, reducing the extent of overlap between the valence band and the reduced state in the solution. Thus the rate constant of hole transferring to the solution decreases, which is consistent with the observation of photocurrent. In other words, specific adsorption of Cl- on the electrode causes its energy band edges to move towards positive value, and also to lower the photocurrent. So in the presence of Cl-, it should produce less OH? radicals and decrease the decay rate of ions, which is contrast to the experimental results as stated above. Therefore, it can rule out that the enhanced degradation rate of is due to an increase in OH? radicals.

Fig.3 Effect of NaCl concentration on photocurrent of TO-TiO2 electrode at +1.0 V: 1 Without NaCl; 2 0.001 mol/L NaCl; 3 0.005 mol/L NaCl; 4 0.01 mol/L NaCl; 5 0.1 mol/L NaCl
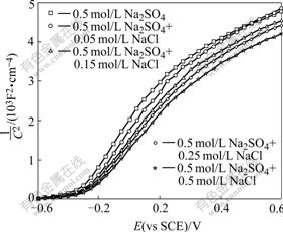
Fig.4 Mott-Schottky plots of TO-TiO2 electrode in 0.5 mol/L Na2SO4 (pH 4.0) solutions with different NaCl concentrations
Although the rate of hole transferring to the solution to form OH? radicals is depressed in the presence of Cl-, as shown by Eqns.(5)-(8), Cl- can also scavenge hole to form active species available for the decay of . The oxidation power of active chlorine is not stronger than that of OH? radicals, but its life is much longer manifesting itself by that it can be detected even after finishing the experiments for several hours. This suggests that active chlorine can diffuse into the bulk solution to react with, differing from OH? radicals that usually only survive in the region of interface. With Cl- in the solution, the active species available to take part in the oxidation rate of do not decrease in the experiments.
3.5 Comparison of different photoanodes
The photoactivity of SG-TiO2 and TO-TiO2 electrode was compared. It can be seen in Fig.5 that in the absence of Cl-, the degradation rate of on the former is faster than that on the latter, although the photocurrent of the former (about 5 mA) is less than that of the latter (25 mA). However, as Cl- is added into the solution, higher rates both for the degradation and active chlorine generation are observed on the TO-TiO2 electrode at the same applied potential.
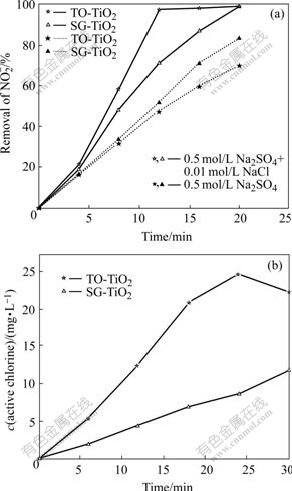
Fig.5 Photoelectrochemical degradation of on two kinds of TiO2 electrode at +1.0 V in 0.5 mol/L Na2SO4 solutions (pH 4.7) (a) and photo-generating active chlorine on TiO2 electrodes at +1.0V in 0.5 mol/L NaCl+0.5 mol/L Na2SO4 solutions (pH 4.7) (b)
Many researchers found an advantage of anatase over the rutile modification of TiO2 in terms of photoactivity[2,14]. Note that the crystalline type was rutile for the TO-TiO2 but anatase for the SG-TiO2. This concept was also well demonstrated by the fact that the anatase SG-TiO2 electrode had a higher photocatalytic activity than rutile TO-TiO2 in the solution without Cl- ion as indicated above, though the photocurrent of the former was less than that of the latter, which may be a reason that SG-TiO2 electrode had a larger surface area than the TO-TiO2 electrode and could therefore be assumed to be more effective. Another more important reason was that the SG-TiO2 could provide more active species available for the PEC degradation of targets while a larger fraction of the photocurrent was used for water oxidation in the case of TO-TiO2[14]. In fact, in our previous work, it was also found that compared with the SG-TiO2 electrode, the TO-TiO2 electrode had a higher light to electricity conversion efficiency, and thus it turned out to be more suitable for the production of H2O2 rather than the degradation of organic pollutant [14].
It is generally accepted that increasing anodic bias potential over TiO2 electrode mainly leads to the increase in space charge layer and band bending, thus it promotes the separation of photogenerated carriers and interface charge transfer rate of the semiconductor/solution, and causes the increase in the photocurrent and quantity of active species, consequently, the degradation rate increases. The particles on the SG-TiO2 electrode were small, which may not form an appreciable depletion layer. In addition, it was porous, thus the immersed electrolyte could screen the electric field. Hence the benefits of electric field enhancement in photocurrent are reduced and there is a significant residual recombination of electrons and holes. In other words, the PEC rate on the TO-TiO2 electrode can be improved by exerting more broad range of anodic potential. So the applied potential of +1.0 V may play an important part in the production of active chlorine. In conclusion, with an applied potential the photoactivity of TO-TiO2 electrode in the degradation of is comparable even superior to that of the anatase TiO2 electrode, and it is also more suitable for the generation of active chlorine.
4 Conclusions
1) can be photoelectron-catalytically oxidized more effectively on the TiO2 film electrode in the solution containing Cl- ion. Active chlorine is detected synchronously in the reaction.
2) Low pH and high concentration of Cl- solutions are beneficial to the degradation rate of . In situ photoelectro-chemical generated active chlorine that can diffuse into the bulk solution, is responsible for the enhancement of degradation rate of with Cl-.
3) The rutile TO-TiO2 film electrode has an advantage of the anatase TiO2 in terms of photoactivity and active chlorine production. The photoelectro- chemical process with in situ generated active chlorine will be an attractive method in the treatment of wastewater.
References
[1] FOX M A, DULAY M T. Heterogeneous photocatalysis [J]. Chem Rev, 1993, 93: 341-357.
[2] HOFFMANN M R, MARTIN S T, WONGYONG C, BAHNEMANN D W. Environmental applications of semiconductor photocatalysis [J]. Chem Rev, 1995, 95: 69-96.
[3] ANDREW M, STEPHEN L H. An overview of semiconductor photocatalysis [J]. J Photochem Photobiol A: Chem, 1997, 108: 1-35.
[4] LUO J, HEPEL M. PEC degradation of naphthol blue black diazo dye on WO3 film electrode [J]. Electrochim Acta, 2001, 46: 2913-2922.
[5] KIM D H, ANDERSON M A. Photoelectrocatalytic degradation of formic acid using a porous TiO2 thin-film electrode [J]. Environ Sci Technol, 1994, 28: 479-403.
[6] KASSIM A, YUSOF N A. Effect of supporting electrolytes in electrochemically-assisted photodegradation of an azo dye [J]. J Photochem Photobiol A: Chem, 2005, 172: 316-321.
[7] SUN C C, CHOU T C. Kinetics of anodic oxidation of nitrite ion using in situ electrogenerated HClO in a NaCl aqueous solution [J]. Ind Eng Chem Res, 1999, 38: 4545-4551.
[8] SHI H X, QU J H, WANG A M, WANG A M, GE J T. Degradation of microcystins in aqueous solution with in situ electrogenerated active chlorine [J]. Chemosphere, 2005, 60: 326-333.
[9] ZANONI M V B, SENE J J, SELCUK H, ANDERSON M A. Photoelectrocatalytic production of active chlorine on nanocrystalline titanium dioxide thin-film electrodes [J]. Environ Sci Technol, 2004, 38: 3203-3208.
[10] SELCUK H, ANDERSON M A. Effect of pH, charge separation and oxygen concentration in photoelectrocatalytic systems: Active chlorine production and chlorate formation [J]. Desalination, 2005, 176: 219-227.
[11] LI G Y, AN T C, CHEN J X, SHENG G Y, FU J M, CHEN F Z, ZHANG S Q, ZHAO H J. Photoelectrocatalytic decontamination of oilfield produced wastewater containing refractory organic pollutants in the presence of high concentration of chloride ions [J]. J Hazard Mater, 2006, 138(2): 392-400.
[12] LENG W H, LIU H, CHENG S A, ZHANG Z, ZHANG J Q, CAO C N. Kinetics of photocatalytic degradation of aniline in water over TiO2 supported on porous nickel [J]. J Photochem Photobiol A: Chem, 2000, 131: 125-132.
[13] LENG W H, ZHANG Z, ZHANG J Q. Photoelectrocatalytic degradation of aniline over rutile TiO2/Ti electrode thermally formed at 600 ℃ [J]. J Mol Catal A: Chem, 2003, 206: 239-252.
[14] LENG W H, ZHU W C, NI J, ZHANG Z, ZHANG J Q, CAO C N. Photoelectrocatalytic destruction of organics using TiO2 as photoanode with simultaneous production of H2O2 at the cathode [J]. Appl Catal A: Gen, 2006, 300: 24-35.
[15] SHI J Y, LENG W H, ZHU W C, ZHANG J Q, CAO C N. Electrochemically assisted photocatalytic oxidation of nitrite over Cr-doped TiO2 under visible light [J]. Chem Eng Technol, 2006, 29: 146-154.
[16] SUN C C, CHOU T C. Electrochemically promoted photocatalytic oxidation of nitrite ion by using rutile form of TiO2/Ti electrode [J]. J Mol Catal A: Chem, 2000, 151: 133-145.
______________________
Foundation item: Projects(20373062; 20107006) supposed by the National Natural Science Foundation of China
Corresponding author: LENG Wen-hua; Tel: +86-571-87952318; Fax: +86-571-87951895; E-mail: lengwh@css.zju.edu.cn
(Edited by LI Xiang-qun)

