文章编号:1004-0609(2011)06-1463-10
基于第一性原理揭示活性元素铪对热障涂层
关键界面的强化机理
江 勇1, 2
(1. 中南大学 材料科学与工程学院,长沙 410083;
2. Materials Department, University of California, Santa Barbara, CA, 93106, USA)
摘 要:本文总结回顾作者近年来从第一性原理出发,以应用广泛的高温热障涂层中γ-Ni(Al)/α-Al2O3关键界面为基础,通过建立合理的热力学模型,结合电子密度泛函计算,针对界面结合强度与温度、原子化学配比和活度等的相关性所开展的一系列理论研究实践。结果表明:在感兴趣的温度区间内(1 300~1 600 K),界面平衡相为富Al相,但靠近理想化学配比相的相界;富Al相界面的结合强度较高,约3倍于理想化学配比相界面的结合强度。杂质S可向界面强烈富集,并严重削弱界面强度的60%~70%;活性元素Hf具有在基体中有效钉扎S、直接参与界面成键和在界面处置换S的3种界面强化效应。
关键词:热障涂层界面;界面结合强度;第一性原理;密度泛函;活性元素
中图分类号:TG132.32 文献标志码:A
Strengthening mechanisms of reactive element Hf on key interface in thermal barrier coating systems based on first-principles
JIANG Yong1, 2
(1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Materials Department, University of California, Santa Barbara, CA, 93106, USA)
Abstract: A systematic strategy was proposed and overviewed to predict the adhesion strength of metal/oxide interfaces, with the technologically-important γ-Ni(Al)/α-Al2O3 interface as an example. The atomistic density functional theory (DFT) calculations were carried out to assess the effects of temperature, interfacial stoichiometry, Al activity, S segregation and Hf doping on the adhesion. Computations of the Al activity in Ni(Al) and the interfacial phase diagram between 1 300 and 1 600 K suggest that the interface phase is Al-riched phase, but close to the boundary with the stoichiometric phase. The Al-riched phase has significantly stronger adhesion than the stoichiometric phase. While S can substantially decrease the adhesion by 60%-70%, alloying with reactive-element Hf substantially improves the adhesion through three strengthening mechanisms: pinning S in bulk Ni(Al), displacing S from its interstitial interfacial sites, and directly enhancing the interfacial bonds.
Key words: thermal barrier coating (TBC); interface adhesion; first-principles density functional theory; reactive element
高可靠性的高温热障涂层系统(Thermal barrier coating systems, TBCs),在新一代航空发动机、燃气轮机和高温固体燃料电池等众多应用领域有着广泛而迫切的需求。自20世纪80年代以来,大量试验研究和使用经验逐步证实,在高温制备或使用过程中,镍基覆衣合金(Bond coat, BC)表面形成的热生长层(Thermally-grown oxide, TGO)氧化铝,可缓解层间热应力和提供一定的抗高温腐蚀作用;但也恰恰在这个覆衣合金/热生长层界面处,最容易发生界面层脱,继而引发整个绝热涂层(YSZ)的直接剥落,被公认为是导致整个材料系统失效的主要原因或方式。该界面(γ-Ni(Al)/α-Al2O3)的结合强度,也因此成为决定高温热障涂层可靠性和寿命的关键。在EVANS等[1]和GLEESON[2]撰写的研究综述中,均对此进行过详尽讨论和深入分析。针对此关键界面展开的进一步实验研究普遍发现,覆衣合金基体中残留的杂质(如硫)有向界面富集/偏析的强烈趋势,并因此弱化界面强度,甚至形成空洞等裂纹源,进而导致氧化层的完全剥 落[3-5],在材料界面学界被专门称为“硫效应”,并从此在很大程度上引导着高温合金的设计和研究方向;活性元素(Hf、Y、Zr、Ce等)在自然界中均存在稳定硫化物,因而在合金中可能具备潜在的钉扎硫的能力,按此思路添加少量这些活性元素后发现,界面层脱均不同程度的缓解[6-8]。针对这些活性元素的作用,从实验研究方面不难总结出两种假设性机理:1) 在基体合金中可能有效钉扎杂质硫,阻滞后者向界面的有害聚集;2) 在界面可能直接参与成键,强化界面的化学结合。但其中所涉及的诸多细节仍然不能确定,也缺乏直接的实验验证[8]。在实际服役工况条件下,这些机理可能单独或同时发生作用,并相互影响(可能相互促进或抑制)。单纯依赖现有的实验方法和检测技术,尚难以系统而全面地认识界面微观结构及其宏观性能的内在联系,从机理上充分揭示杂质或合金元素对界面性能的影响和作用。
随着界面科学自身的不断发展,有关此类复杂界面的一系列基础性问题,也迫切需要从理论上寻找明确答案。比如这类合金/氧化物界面的理论结合强度值;界面强度受杂质、基体合金成分和制备条件等影响和控制的内在机理;获得接近、达到、甚至超过理论界面强度的科学途径和方法(如基体合金的元素优化)等。针对这类基础性问题,本文作者提出应用第一性原理计算研究方法,对该界面的界面强度与生长环境、相结构、元素成分的相关性开展系统性研究,为科学设计基体合金、预测涂层界面寿命和可靠性提供理论基础。
第一性原理计算研究方法的最大特点是从最基本的热力学原理出发,以计算量子力学为手段,对体系能量(焓、熵、自由能)、晶体结构和电子结构等进行无参数的精确计算,结合合理的物理和热力学模型,可对材料的宏观物理性能、化学性能和机械性能等展开直接的理论预测。其计算过程由于不需要引入任何经验性参数或实验数据,计算结果依靠能量准则(或原子间力准则)自我收敛,故可排除一切人为因数的影响,研究结论可以做到自我支持。近20年来,随着并行计算科学和技术的飞速发展,基于第一性原理的计算研究方法,已成为当今材料交叉、前沿学科日渐受到重视的一个主要的,可靠的理论研究手段。
CARLING和CARTER[9]和JARVIS等[10]先期开展了针对热生长层界面的第一性原理计算研究,通过计算比较引入外来元素前后界面结合能的变化,初步确定了各元素的界面强弱化性质。但该小组的研究并未评估各元素的界面偏析能力,也未考虑杂质钉扎的可能性,尤其是对热生长层的热力学特点完全忽视,把合金/热生长层界面当成了普通界面处理,未考虑Ni(Al)中Al活度和温度对界面结构的决定性影响,从而割裂微观界面结构与外部生长环境的相关性,导致其后续有关界面杂质和合金元素的计算和讨论,缺乏合理的说服力。针对这些局限性,本文作者提出分析和把握热生长层界面的热力学特点,运用第一性原理建立合理的热力学模型,通过电子密度泛函计算界面体系总能,全面、系统地研究纯净界面的界面平衡相结构,界面的理论结合强度,元素的界面偏析能力和偏析行为,偏析对界面结构和强度的影响以及Hf在合金基体内钉扎S的能力,特别是在服役高温下的钉扎有效性。
具体而言,本文作者拟开展的计算研究将包括以下几项主要内容:
1) 计算γ-Ni(Al)/α-Al2O3界面相结构与温度和气氛(氧分压或Al活度)的关系,即“界面相图”;
2) 分别计算Hf和S的界面偏析能,并确定可能的偏析路径;
3) 分别计算和比较理想纯净界面和受Hf和S偏析界面的界面脱粘功(Work of separation,Wsep),评估界面偏析对结合强度的影响;
4) 计算Hf-S在基体内的相互作用能,揭示元素钉扎的微观机理,并考察温度对钉扎能力的影响;
所有计算采用第一性原理电子密度泛函通用程序包VASP[11],其中交换关联能采用广义梯度近似,电子与核之间的相互作用采用Vanderbilt超软赝势,平面波展开采用较高的能量截断(400 eV)。有关Al化学活度的计算,采用包含单个Al原子的2×2×2 面心γ-Ni超点阵,布里渊内的能量积分在6×6×6 Monkhorst-Pack(M-P) k-网格中进行, 温度对能量的贡献通过直接超胞法的声子计算和准简谐近似估算[12]。有关界面的计算,采用三明治型的共格γ-Ni(111)/α-Al2O3(0001)界面模型,整个超胞包含8层Ni原子、12层Al原子、6层O原子和至少12?的真空层,布里渊内的积分采用3×3×1 M-P k-网格。有关基体的计算,采用包含单个Hf-S原子对的3×3×3面心γ-Ni的超点阵和2×2×2 M-P k-网格。为获得平衡态结构,驰豫计算采用Hellmann-feymann原子净力收敛判据:20 meV/? (1 eV/?=1.6 nN)。所有计算考虑自璇极化,但暂且忽略声子-电子的相互作用和磁性变化对能量的影响。
1 界面结构与界面强度
1.1 界面平衡相结构的确定
高温镍合金/热生长层界面的形成过程,首先是基体镍基合金中固溶的铝元素,在基体表面得到优先氧化,由于铝的逐渐消耗,临近界面的镍基合金组织可能发生相应转变,由通常的两相(γ/β或γ/γ′)转变成单相(γ),氧化过程也因铝的消耗和氧化层的增厚而逐渐减缓,最终接近或达到热力学平衡(参见图1[13])。与一般第一性原理计算常规界面的方法[9-10, 14-15]不同,本文作者研究的这类界面的结合强度,首先考虑热生长过程中的热力学平衡条件,即从计算界面平衡相图开始。
1.1.1 界面原子结构类型的选取
第一性原理考察界面平衡相结构,从构建界面原子结构类型开始。异型界面两侧晶格常数的差异,导致在界面附近不可避免出现失配位错。然而在界面模拟中,构建一个足够大的异型界面超胞,将这类失配位错“自然”包含在内,将导致极大的计算量。采用的折衷方案是,在γ-Ni/α-Al2O3界面附近晶格中引入相应合理的尽可能小的变形量,以形成理想共格的界面原子结构模型。在计算对应的界面能和脱粘功(即界面强度)时,通过界面和两个脱粘表面体之间的能量相消,最大程度消除表面体内的应变对界面能/界面强度在量值上的干扰。

图1 典型多层热障涂层系统的界面体系组成及各组元层的基本功能[13]
Fig.1 Cross sections of actual multi-layer thermal barrier system indicating functionalities of each of layers[13]
图2和表1所示为3种被考察的界面原子结构类型的构建关系,分别代表3种典型的晶体取向和共格应变量的组合,其中在具有γ-Ni(111)( )// α-Al2O3(0001) (1×1)取向关系的结构中(即型号Ⅰ和Ⅱ),金属Ni一侧均承受拉伸应变,与已有的类似界面(Nb/Al2O3)的原子结构高分辨电镜(HRTEM)观察[6]较符合(参见图3,照片显示Nb/Al2O3界面处的失配位错分布在金属Nb一侧,距离界面有若干晶面层间距,并在Nb中产生局部拉伸应变。假定Ni/Al2O3界面的失配位错存在类似效果)。尤其是型号Ⅱ,因对应的共格变形相对最小,故在后续计算中采用。
)// α-Al2O3(0001) (1×1)取向关系的结构中(即型号Ⅰ和Ⅱ),金属Ni一侧均承受拉伸应变,与已有的类似界面(Nb/Al2O3)的原子结构高分辨电镜(HRTEM)观察[6]较符合(参见图3,照片显示Nb/Al2O3界面处的失配位错分布在金属Nb一侧,距离界面有若干晶面层间距,并在Nb中产生局部拉伸应变。假定Ni/Al2O3界面的失配位错存在类似效果)。尤其是型号Ⅱ,因对应的共格变形相对最小,故在后续计算中采用。
1.1.2 界面原子化学配比的影响
合金/热生长层界面的平衡相结构,由实际热生长环境条件(如温度和化学反应气氛)决定,其中化学气氛可由氧分压或合金中的Al活度表征,它直接决定着参与界面化学反应的各元素原子的相对含量,即界面附近的原子化学配比(Stoichiometry)。参考表面终端(Termination)的概念,设想热生长化学环境条件下可能得到的3种典型的界面原子化学配比类型,即理想化学配比(Ni/Al2O3-)、富Al(Ni/AlAl2O3-)和富O型(Ni/OAl2O3-)[17],分别计算它们对应的界面能,通过界面能与温度和Al活度之间的热力学关系,可绘制出界面相结构对应于Al活度的“界面平衡相图”。
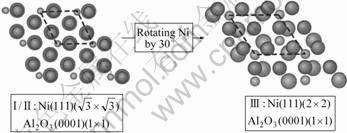
图2 3种典型界面结构类型图
Fig.2 Three commensuration structures for atomistic γ-Ni/ α-Al2O3 (Ni, Al, O atoms are represented in dark blue, green and red, respectively. Colour figures available on line)
表1 3种典型界面结构类型参数
Table 1 Parameters of three commensuration types for atomistic γ-Ni/α-Al2O3

图3 含失配位错的Nb/Al2O3界面原子结构的高分辨TEM像[16]
Fig.3 HRTEM image of Nb/α-Al2O3 interface containing misfit edge dislocation showing stand-off and tensile strain in Nb along atomic layers
计算结果表明,相比于晶体取向和共格应变,界面原子化学配比是决定界面能的主要因数。从图4可以看到,界面结构类型不同,但原子化学配比类型相同,各界面强度(以界面脱粘功衡量,这里暂且假设脱粘总是在Ni和Al2O3界面原子层之间发生)相差最大不超过50%;而界面结构类型相同,但原子化学配比不同,界面强度相差则可达数倍以上。以在后续计算中被采用的型号Ⅱ界面为例,其富Al和富O型界面相的界面强度,分别是其理想化学配比界面相的4倍和6倍。这种理论结合强度极高的界面相,对应所需的热力学生长条件,可以从计算得到的界面“相图”上确定。

图4 3种典型界面原子结构类型的不同化学配比对应的界面强度
Fig.4 Interfacial adhesion strength (evaluated as work of separation) as function of interfacial stoichiometry for different commensurate interface types
1.1.3 Al活度与界面平衡相图的关系
根据界面能的定义,不难推导出热力学平衡条件下Ni/Al2O3界面能的表达式[18]
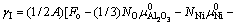
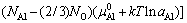 (1)
(1)
式中:A是界面的横截面积;Fo是整个超胞的Helmholtz自由能;上标‘0’表示各组元的纯净态;Ni和μi分别是各组元元素i的原子个数和化学势(单位自由能)。由于前四项自由能之间存在大量的相消,温度对界面能的影响主要体现在最后一项,即 ,其中
,其中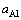 是γ-Ni(Al)基体的Al活度,是建立界面与基体之间热力学平衡的关键物理量,它本身是温度和浓度(xAl)的函数。通过热力学理论进一步推导,可获得如下关系[10]
是γ-Ni(Al)基体的Al活度,是建立界面与基体之间热力学平衡的关键物理量,它本身是温度和浓度(xAl)的函数。通过热力学理论进一步推导,可获得如下关系[10]
 (2a)
(2a)
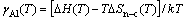 (2b)
(2b)
式中: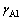 是基体Ni(Al)合金中的Al活度因子,在低浓度(xAl≤10%~15%)下一般可认为与浓度无关,即
是基体Ni(Al)合金中的Al活度因子,在低浓度(xAl≤10%~15%)下一般可认为与浓度无关,即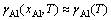 。DH和DSn-c分别对应于单个Al原子从纯净单质固溶到γ-Ni(Al)基体中所发生的焓变和非形熵变,均可以通过密度泛函计算得到[10]。
。DH和DSn-c分别对应于单个Al原子从纯净单质固溶到γ-Ni(Al)基体中所发生的焓变和非形熵变,均可以通过密度泛函计算得到[10]。
图5所示为计算得到的Al活度(或活度因子)随温度的变化。与相关实验值比较,发现计算值与实验在高温区间普遍吻合较好,但在较低温度(980 K)下与实验测量值偏差在4个数量级左右。一般认为这主要是由于该温度接近Al熔点,通过蒸汽分压测得的实验值误差太大,可信度不高。相比之下理论计算从热力学原理出发,不受实验条件局限,可涵盖较广泛的温度区间,所得预测应更可靠。无论如何,本研究将主要针对高温区间T为1 300~1 600 K。
针对不同的界面相结构,计算任意给定温度下界面能与Al活度的对应关系,就可以得到“界面平衡相图”的一个等温界面。图6所示为通过式(1)和(2)计算得到T=1 300 K下的等温截面。根据实验已知热生 长界面附近因Al择优氧化而消耗后的大致浓度范围, 1%≤xAl≤15%,结合计算得到该温度下的Al活度因子,可以估计界面附近Ni(Al)一侧Al元素的大致活度范围(图6中以橙色柱标示)。借助图6所示的等温截面“相图”,可以判断该温度下界面平衡相是富Al相,但靠近理想化学配比相的相界。由图6还可知,在该温度下,随着Al活度不断降低(即富氧气氛,对应氧分压不断升高),界面平衡相将逐步过渡为理想化学配比相、富氧相、复合氧化物NiAl2O4、直至NiO;在高Al活度(即乏氧气氛)条件下,界面可直接生成金属间化合物Ni3Al。其中Al活度和环境中的氧分压之间存在一定的换算关系,可以由合金表面反应:Al(s)+O2 (g) = Al2O3(s)的热力学平衡条件推导出来[18]。
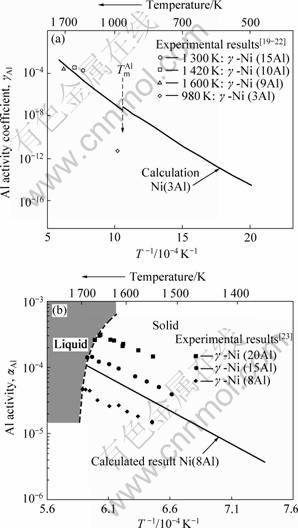
图5 计算预测的Al活度(系数)随温度的变化
Fig.5 Al activity (coefficient) for γ-Ni(Al) and comparison with measurements

图6 计算得到的界面平衡相图中T=1 300 K的等温截面
Fig.6 Interfacial phase diagram calculated at 1 300 K
1.2 界面偏析与界面强度
根据界面附近Al浓度的大致范围,借助计算得到的“界面相图”可以判断,T =1 300 K左右,界面平衡相应为富Al相,但也接近理想化学配比相的相界。在计算有关杂质和活性元素的界面偏析时,为谨慎起见,将同时考虑这两种界面相结构(即富Al相和理想化学配比相)上的偏析行为,并比较其对界面强度的影响。
1.2.1 界面偏析能和偏析路径的关系
如前所述,同样的基体,在不同生长条件(温度和气氛)下,得到的界面平衡相可能不同,因此活性元素和杂质的界面偏析能力也会相应不同,从而直接影响到它们对界面的影响和作用。界面偏析能力可以用界面偏析能来计量。首先计算两类纯净界面对应的总能量,然后从基体中引入活性元素和杂质到纯净界面的不同位置,计算前后总能的变化,即可得该元素针对某类界面某个特定位置的界面偏析能。正值的界面偏析能代表从基体向界面的偏析趋势(反之亦然)。比较同一元素在界面不同位置上的界面偏析能,可以确定该元素的偏析路径。比较界面上同一位置不同元素的界面偏析能,可以确定元素之间的偏析优先权。
图7所示为计算得到的界面偏析趋势。此处的界面偏析度以Ni(111)层上每Ni原子为单位。括号中的数字代表各偏析进程对应的界面偏析能(单位:eV/atom)。1) 对于理想化学配比相的界面,与实验发现一致,杂质S有着强烈的界面偏析趋势。在界面的填隙位(SI)和Ni置换位(SNi),杂质S相应的界面偏析能分别高达1.55和1.24 eV/atom。不过,活性元素Hf有相对更大的偏析趋势,在Al置换位(HfAl)、界面填隙位(HfI)和Ni置换位(HfNi),Hf相应的界面偏析能分别达到1.74、1.79、和1.29 eV/atom。2) 对于富Al相的界面,S的界面偏析驱势大大减少,在界面Ni置换位(SNi)和填隙位(SI)上,S的界面偏析能仅分别为0.42 和0.18 eV/atom。相对而言,Hf依然具有稍大的偏析趋势,在Al置换位(HfAl)上偏析能为0.66 eV/atom。显而易见,在两类界面上,Hf的界面偏析均优先于杂质S的,而杂质S能否在界面偏析,将不得不取决于先期抵达界面的Hf的偏析行为。以理想化学配比相的界面(图7(b))为例,如果Hf的偏析事先不存在,偏析到界面的杂质S可以继占据Ni置换位(SNi)之后,再占据界面上的填隙位(SI),体系总能进一步降低0.67 ev/atom。如果界面已有偏析Hf存在,由于Hf-S在界面上仍存在较强的相互作用(强于Hf-Hf在界面上的相互作用),杂质硫的偏析仍然可以继续进行,在界面上出现所谓Hf和S的“共同偏析”,体系总能大幅度降低。从图7(a)可以看到,类似的共同偏析也可以在富Al相的界面上出现。界面偏析(包括共同偏析)对界面强度的影响,可以通过直接计算相应的界面脱粘功,定量获知。
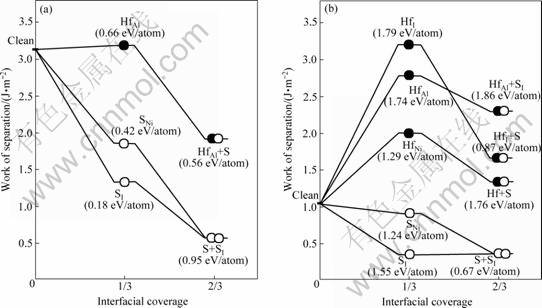
图7 富Al相界面和理想化学配比相界面的界面脱粘功(Wsep)随界面偏析度的变化规律
Fig.7 Work of separation, Wsep, as function of interfacial coverage for Al-rich(a) and stoichiometric interfaces(b)
1.2.2 元素偏析对界面强度的影响
针对每一种可能的偏析路径,可以通过一系列总能的计算,在对应的界面结构中寻找结合强度最弱的原子层,计算从该处分离界面所需的能量,即界面脱粘功(Wsep),并以此作为该界面结合强度的度量。显然,比较偏析前后界面结构的变化,计算相应所需的界面脱粘功,可以定量预测该元素偏析对界面结合强度的影响。
有关界面脱粘功的计算结果如图7和8所示,其中图8下方为相应的界面俯视图,以显示界面共格关系以及偏析元素在界面上的原子位置。对于感兴趣的界面,可以绘制出相应的基态价电子的密度分布云图(见图9),结合计算得到的界面脱粘功,从而能够定性地分析界面附近电子成键的特点,以及合金元素和杂质偏析对界面化学成键的不同影响。
作为热力学平衡相的富Al相的纯净界面其实具有相当高的界面结合强度(Wsep=3.13 J/m2),约3倍于理想化学配比相界面的。对比图9(a)和(b)可知,这是因为富Al相界面上形成有很强Ni―Al金属键(见图9(a)),远强于理想化学配比相界面上的Ni―O键(共 价―离子混和键,见图9(b))。在这两类界面上,特别是在理想化学配比相界面上,杂质硫对界面强度危害极大,可降低界面脱粘功达60%~70% (见图7和8)。对比图9(b)和(d)可知,由于硫的界面偏析,界面上原本还算较强的Ni―O被较弱的共价键S―O和更弱的离子键S―Al替换(见图9(d)),导致界面弱化。同样值得注意的是,活性元素Hf强化界面的作用,在相对较弱的理想化学配比相界面上,表现得尤为明显(见图7(b)和8)。特别是当Hf偏析到填隙位置(HfI),可使Wsep提升约300%。仔细观察计算得到的基态界面原子结构可知,实际上源自于界面原子结构的改变:Hf在理想化学配比相界面的填隙位上的偏析(HfI),对界面微观结构发生影响,使其恰巧复制了Hf在富Al相界面上的Al置换位偏析(HfAl)的界面原子结构。对应的界面电子密度云图(见图9(c)) 清楚显示,此时的Hf 能够同时与界面两侧的Ni 和O形成新键,而且新键的键能远强于图9(b)中Ni―O的,接近图9(a)中 Ni―Al金属键的,从而使原本较弱的理想化学配比相界面的结合强度得到近3倍的提升,达到富Al相纯净界面的理论结合强度。这充分证实了实验研究推断的Hf直接参与界面成键,从而强化界面的机理假说。同时,从图7中也可以清楚看到,Hf对界面的强化作用,容易遭后续偏析的S破坏而消减,因此对于这两类界面,Hf和S的共同偏析需要尽可能的避免。只有设法将S有效地钉扎在基体中,避免其在界面的偏析,才有可能最大限度的发挥Hf的强化作用。换句话说,在Ni基体中能否有效钉扎S,是决定Hf能否充分发挥其界面强化效用的重要前提。
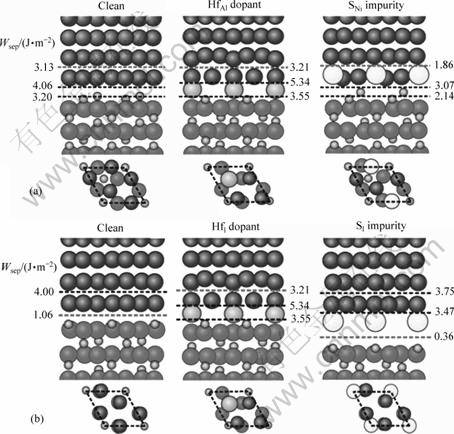
图8 界面偏析对(a)富Al相界面和(b)理想化学配比相界面的结合强度的影响
Fig.8 Segregation structures and Wsep of Al-rich(a) and stoichiometric interface(b) at 1/3 monolayer coverage of Hf and S (Ni, Al, O, Hf, S atoms are represented in dark blue, green, red, light blue and yellow, respectively, colour figures available on line)
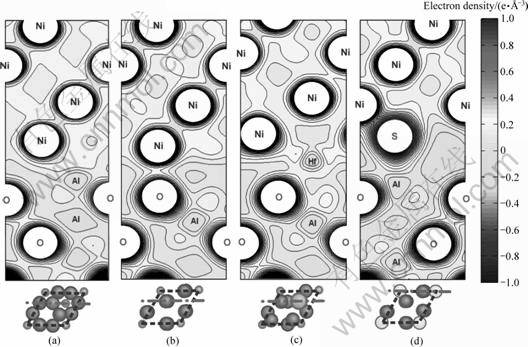
图9 界面基态电子密度云图
Fig.9 Electron density contours: (a) Al-riched interface; (b) Stoichiometric interface; (c) Stoichiometric interface containing segregated interstitial Hf; (d) Stoichiometric interface containing segregated interstitial S (Dash-dot lines at bottom correspond to positions of contour planes in top-views)
2 基体内的杂质钉扎
2.1 钉扎能与温度的影响
添加活性元素(如Hf),是考虑到自然界中存在其稳定的硫化物,因此有可能在基体合金中与S积极反应,从而通过钉扎S,抑制其有害的界面偏析。但这种钉扎效应在Ni基体环境中是否真正存在,特别是高温环境下这种钉扎是否真正有效,无论在实验还是理论研究,还缺乏直接证据。
为证实Hf-S原子对在γ-Ni基体中的相互作用的有无和强弱,本文作者进一步提出,可以通过电子密度泛函方法,分别计算Hf-S原子对在γ-Ni(FCC)晶格中的最近邻(2.5 ?)距离和理论上无限远距离上的系统总能,比较两个总能之差, 可定量反映Hf-S原子对之间相互作用的大小。如果前者的总能较高,即总能之差为正,Hf-S原子对在γ-Ni基体中就趋向分裂;反之,如果后者的总能较高,即总能之差为负,Hf-S原子对在γ-Ni基体中就趋向稳定,表明S能够被Hf有效钉扎,且总能之差越负,钉扎能力越强。实际计算中,选择以FCC晶格中的第五近邻距离作为无限远近似(约8.5 ?),可以确保此时两个原子间距足够远,相互作用可忽略不计。
有关计算结果可见图10(虚线表示0 K下计算得到的作用能,可反映较低温度下的钉扎能力),计算充分考虑温度对晶格的振动熵的贡献(即热声子),以及热电子效应[24]。从图10中可看到,在0 K下,不存在任何温度效应时,Ni基体中的Hf和S能形成稳定的原子对,且相互作用较强,约0.27 eV/atom。随着温度的升高,如果只单独考虑Ni晶格的热膨胀效应,Hf-S原子对间距以及其与基体Ni原子的间距增 加,有利于Hf-S原子对结合能的进一步增强,但高温下同时存在晶格热振动(声子)和电子的热效应,这两部分的作用较大程度地削弱了Hf-S原子对之间的相互作用。仍然,计算表明在感兴趣的整个高温区间 (1 300~1 600 K),Hf 都能较强地钉扎S,两者之间的相互作用能在 0.17~0.20 eV左右。
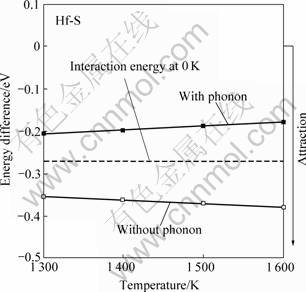
图10 高温下γ-Ni基体中Hf-S原子对的相互作用能随温度的变化
Fig.10 Change of calculated interaction energies of Hf-S and Pt-S pairs in Ni with temperature at high temperatures (Dashed lines refer to interaction energies calculated at 0 K, reflecting low-temperature pair attraction)
2.2 Hf-S钉扎的微观机理
为有效揭示Ni基体中Hf存在较强钉扎杂质S的作用机理,计算Hf-S原子对在Ni基体中的价电子局域化函数分布(Electron localization function, ELF[25])和差分电荷密度。图11(a)清楚显示,在Ni金属的均匀电子密度背景衬托下,Hf-S原子对有明显的电子局域化:电子密度主要在S附近且沿Hf方向相对集 中。图11(b)清楚显示Hf-S原子对的相互作用主要是离子键性质,并混合少量的共价成分:在Ni基体中,Hf的价电荷少部分向周围Ni,大部分向S发生迁移, 而S从周围Ni,特别从Hf获得价电荷,与图11(a)中电子在S附近局域增强相对应。电子态密度分析也证实有微弱的Hf(5d,6s)-S(3p)轨道杂化。这种价电荷的充分迁移,是决定Hf-S原子对在Ni基体中能够具备较强亲和力的微观机制。至此,活性元素Hf在γ-Ni基体中钉扎杂质S的有效性,经计算得到充分证实。
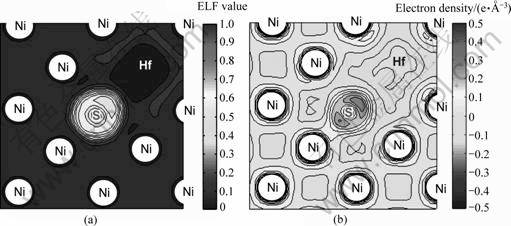
图11 显示S在基体中能有效捕获电子局域化函数等势图和显示价电荷从Hf到S的迁移差分电荷等势图
Fig.11 Electron localization function contours showing S effectively gathering electrons in Ni matrix(a) and differential valence charge densities of Hf-S pair in Ni showing significant charge transfer from Hf to S(b)
3 结论
1) 合金/热生长层界面的平衡相结构由生长气氛(Al活度或O分压)和环境温度决定。通过本方法计算得到合金中的Al活度和界面平衡相图,可以理论预测任意给定生长条件下对应得到的界面平衡相结构。
2) 在感兴趣的温度区间内(1 300~1 600 K),界面平衡相为富Al相,但靠近理想化学配比相的相界。偏离热平衡条件下,两种界面相可能共存。
3) 取决于具体的界面相结构,γ-Ni/α-Al2O3理想纯净界面的理论结合强度(以界面脱粘功Wsep衡量)为1~3 J/m2。其中富Al相界面结合强度较高,约3倍于理想化学配比相界面的结合强度。
4) 对于可能共存的两种γ-Ni/α-Al2O3界面相,活性元素Hf和杂质S均存在较强的界面偏聚能力。
5) 即便是少量S偏析到界面上,可立即降低界面结合强度60%~70%。
6) 作为合金元素添加的Hf,其界面强化机理主要包括在Ni基体内有效钉扎杂质S,从而抑制后者的有害界面偏析;直接参与γ-Ni/α-Al2O3界面的化学成键;或在界面处置换偏析杂质S。
7) 即便是高温下(1 300~1 600 K),Hf也能够在Ni基体中有效钉扎S。一旦S的有害界面偏析被完全抑制,偏析到界面的Hf可充分强化界面。尤其是对结合强度较低的理想配比界面相,可提高界面强度300%,使其接近和达到理想纯净富Al相界面的理论结合强度(约3 J/m2)。
REFERENCES
[1] EVANS A G, MUMM D R, HUTCHINSON J W, MEIER G H, PETTIT F S. Mechanisms controlling the durability of thermal barrier coatings[J]. Progress in Materials Science, 2001, 46(5): 505-553。
[2] GLEESON B. Thermal barrier coatings for aeroengine applications[J]. Journal of Propulsion and Power, 2006, 22(2): 375-383.
[3] SMIALEK J L, JAYNE D T, SCHAEFFER J C, MURPHY W H. Effects of hydrogen annealing, sulfur segregation and diffusion on the cyclic oxidation resistance of superalloys: A review[J]. Thin Solid Films, 1994, 253(1/2): 285-292
[4] GAUDETTE F G, SURESH S, EVANS A G. Effects of sulfur on the fatigue and fracture resistance of interfaces between gamma-Ni(Cr) and alpha-Al2O3[J]. Metall Trans A, 2000, 31(8): 1977-1983.
[5] KIELY J D, YEH T, BONNELL D A. Evidence for the segregation of sulfur to Ni-alumina interfaces[J]. Surf Sci, 1997, 393(1/3): L126-130.
[6] FUNKENBUSCH A W, SMEGGIL J G, BORNSTEIN N S. Reactive element-sulfur interaction and oxide scale adherence[J]. Metall Trans A, 1985, 16(6): 1164-1166,
[7] KHANNA A S, WASSERFUHR C, QUADAKKERS W J, NICKEL H. Addition of ytterbium, cerium, and hafnium to combat the deleterious effect of Sulfur impurity during oxidation of an Ni-Cr-Al alloy[J]. Mater Sci Eng A, 1989, 120(1):185-191.
[8] HOU P Y. Segregation phenomena at thermally grown Al2O3/alloy interfaces[J]. Annual Review of Materials Research, 2008, 38: 275-298.
[9] CARLING K M, CARTER E A. Effects of segregating elements on the adhesive strength and structure of the α-Al2O3/β-NiAl interface[J]. Acta Materialia, 2007, 55: 2791-2830.
[10] JARVIS E A, CHRISTENSEN A, CARTER E A. Weak bonding of alumina coatings on Ni (111)[J]. Surface Science, 2001, 487(1/3): 55-76.
[11] KRESSE G, FURTHM?LLER J. Vasp manual. http://cms.mpi.univie.ac.at/ vasp/vasp/vasp.html
[12] JIANG Y, SMITH J R, EVANS A G. Temperature dependence of the activity of Al in dilute Ni(Al) solid solution[J]. Physical Review B, 2006, 74(22): 224110-1.
[13] EVANS A G, CLARKE D R, LEVI C G. The influence of oxides on the performance of advanced gas turbines[J]. J Euro Ceram Soc, 2008, 28(7): 1405-1419.
[14] 胡壮麒, 彭 平, 刘 轶, 金 涛, 孙晓峰, 管恒荣. 镍基合金中γ′相界面的强化设计[J]. 金属学报, 2002,38(11): 1121-1126.
HU Zhuang-qi, PENG Ping, LIU Yi, JIN Tao, SUN Xiao-feng, GUAN Heng-rong. Design of γ' phase interface strengthening of Nickel-base superalloy[J]. Acta Metallurgica Sinica, 2002,38(11): 1121-1126.
[15] 尚家香, 王福合, 徐惠彬. Co对ZrO2/Ni热障涂层界面结合的影响[J]. 北京航空航天大学学报, 2004, 30(10): 958-961.
SHANG Jia-xiang, WANG Fu-he, XU Hui-bin. Effect of Cr on ZrO2/Ni thermal barrier coatings interface cohesion[J]. Journal of Beijing University of Aeronautics and Astronautics, 2004, 30(10): 958-961.
[16] VITEK V, GUTEKUNST G, MAYER J, RUHLE M. Atomic-structure of misfit dislocations in metal-ceramic interfaces[J]. Philosophical Magazine A, 1995, 71(6): 1219-1239.
[17] SMITH J R, JIANG Y, EVANS A G. Adhesion of the gamma-Ni(Al)/alpha-Al2O3 interface[J]. International Journal of Materials Research, 2007, 98(12): 1214-1221.
[18] ZHANG W, SMITH J R, EVANS A G. The connection between ab initio calculations and interface adhesion measurements on metal/oxide systems: Ni/Al2O3 and Cu/Al2O3[J]. Acta Materialia, 2002, 50(15): 3803-3816.
[19] ESKOV V M, SAMOKHVAL V V, VECHER A A. Thermodynamic properties of Al-Ni hard alloys[J]. Russ Metall, 1974, 2: 118-119.
[20] STEINER A, KOMAREK K L. Thermodynamic activities of solid nickel-aluminum alloys[J]. Trans Metall Soc AIME, 1964, 230(4): 786-788.
[21] OFORKA N C. Thermodynamics of nickel-aluminum alloys[J]. Indian J Chem A, 1986, 25(11): 1027-1029.
[22] HILPERT K, MILLER M, GERADS H, NICKEL H K. Thermodynamic study of the liquid and solid alloys of the nickel-rich part of the Al-Ni phase-diagram including the AlNi3 phase[J]. Ber. Bunsenges, Phys Chem, 1990, 94(1): 40-47.
[23] COPLAND E. Dissolved oxygen and the partial thermodynamic properties of γ′-Ni3Al+β-NiAl alloys[J]. Scripta Materialia, 2007, 57(1): 21-24.
[24] JIANG Y, LIU R. Gettering of S in Ni from first-principles[J]. Scripta Materialia, 2010, 62(10): 782-785.
[25] BECKER A D, EDGECOMBE K E N, A simple measure of electron localization in atomic and molecular systems[J]. Journal of Chemical Physics, 1990, 92(9): 5397-5403.
(编辑 李艳红)
基金项目:美国空军研究室项目(FA9550-05-C-0039)
收稿日期:2010-08-11;修订日期:2010-10-28
通信作者:江 勇,研究员,博士;电话:0731-88830450;传真:0731-88876692;E-mail: yjiang@csu.edu.cn

