J. Cent. South Univ. Technol. (2011) 18: 1883-1890
DOI: 10.1007/s11771-011-0918-9
Adsorption of NO and NH3 over CuO/��-Al2O3 catalyst
ZHAO Qing-sen(����ɭ)1, SUN Lu-shi(��·ʯ)2, LIU Yong(����)1,
SU Sheng(��ʤ)2, XIANG Jun(���)2, HU Song(����)2
1. Suzhou Nuclear Power Research Institute, Suzhou 215004, China;
2. State Key Laboratory of Coal Combustion, Huazhong University of Science and Technology,Wuhan 430074, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2011
Abstract: The selective catalytic reduction reaction belongs to the gas-solid multiphase reaction, and the adsorption of NH3 and NO on CuO/��-Al2O3 catalysts plays an important role in the reaction. Performance of the CuO/��-Al2O3 catalysts was explored in a fixed bed adsorption system. The catalysts maintain nearly 100% NO conversion efficiency at 350 ��C. Comprehensive tests were carried out to study the adsorption behavior of NH3 and NO over the catalysts. The desorption experiments prove that NH3 and NO are adsorbed on CuO/��-Al2O3 catalysts. The adsorption behaviors of NH3 and NO were also studied with the in-situ diffusion reflectance infrared Fourier transform spectroscopy methods. The results show that NH3 could be strongly adsorbed on the catalysts, resulting in coordinated NH3 and  NO adsorption leads to the formation of bridging bidentate nitrate, chelating bidentate nitrate, and chelating nitro. The interaction of NH3 and NO molecules with the Cu2+ present on the CuAl2O4 (100) surface was investigated by using a periodic density functional theory. The results show that the adsorption of all the molecules on the Cu2+ site is energetically favorable, whereas NO bound is stronger than that of NH3 with the adsorption site, and key information about the structural and energetic properties was also addressed.
NO adsorption leads to the formation of bridging bidentate nitrate, chelating bidentate nitrate, and chelating nitro. The interaction of NH3 and NO molecules with the Cu2+ present on the CuAl2O4 (100) surface was investigated by using a periodic density functional theory. The results show that the adsorption of all the molecules on the Cu2+ site is energetically favorable, whereas NO bound is stronger than that of NH3 with the adsorption site, and key information about the structural and energetic properties was also addressed.
Key words: CuO/��-Al2O3; NH3; NO; adsorption; diffusion reflectance infrared Fourier transform spectroscopy; density functional theory
1 Introduction
Nitrogen oxides (NO, NO2, and N2O) are mainly produced during the combustion of fuels from mobile and stationary sources. Such combustion results in serious air pollution by the formation of acid rain, photochemical smog, and the greenhouse effect. In recent years, many methods have been used to reduce the emission of nitrogen oxide. Catalytic technologies are attractive because of their low cost and high efficiency.
The CuO/��-Al2O3 catalyst has been applied in fixed-, moving-, and fluidized-bed reactors [1-3]. Our previous studies [4] have shown that this catalyst maintains more than 90% NO reduction efficiency with NH3 at a wide temperature range of 250-400 ��C. The selective catalytic reduction (SCR) reaction belongs to the gas-solid multiphase reaction, and the adsorption of NH3 and NO plays an important role in the reaction. CENTI et al [1] found that NO could be adsorbed on the catalyst surface during the SCR process, and they proposed several reaction paths of SCR between the NH3 and adsorbed NO. RAMIS et al [5-6] investigated the SCR mechanism over CuO/TiO2 at 200-250 ��C by FTIR. They found that the adsorbed NH3 was activated through H abstraction (partial oxidation) to form NH2 (amide), which then reacted with the gas-phase NO to form a nitrosamide (NH2NO) species, an intermediate that was decomposed into N2 and H2O. This mechanism was termed the amide- nitrosamide (NH2-NH2NO) mechanism. LIU et al [7] also pointed out that the SCR activity was determined by the amount of NH3 adsorbed on the surface and the H-abstraction extent of NH3(ads). Based on these viewpoints, it can be concluded that NH3 could be adsorbed on the CuO/��-Al2O3 catalysts, but the adsorbed and oxidized form of NH3 still needs to be further studied, and there have long been controversies and doubts about the adsorption and adsorption effect of NO.
Adsorption properties of NH3, NO2 and NO molecules on CuO/��-Al2O3 catalysts surface are closely related to the crystalline structure. CuO/��-Al2O3 catalysts calcined at high temperatures will produce CuAl2O4 phase. Cu2+ in the CuAl2O4 crystal surface is the catalytic active site during the SCR reaction [8-11]. SHIMIZU and his co-workers [10-11] investigated the reaction mechanism of catalytic reduction of NO with C3H6 using the CuAl2O4 catalyst, as the active component of Cu2+ played an important role in the chemical reactions (the high catalytic activity and good selectivity were both due to the Cu2+). However, the adsorption capacity of NH3 and NO on the Cu2+ site of CuAl2O4, and the charge transfer between the adsorbates and CuAl2O4 surface, as well as the relation of the adsorption capacity with the charge transfer, need to be studied further.
Therefore, the desorption performances of the CuO/��-Al2O3 catalysts are studied in a fixed-flow reactor with temperature-programmed desorption (TPD) method. To obtain more information about the adsorption of NH3 and NO in this system, the diffusion reflectance infrared Fourier transform spectroscopy (DRIFTS) and the density functional theory (DFT) methods are carried out.
2 Experimental
2.1 Desorption experiment
Spherical CuO/��-Al2O3 granular catalysts were prepared by sol-gel method, as described in Refs.[4, 12]. In this work, two catalysts are denoted as Cu8 and Cu20, respectively. The mass fractions of Cu for Cu8 and Cu20 are 8% and 20%, respectively.
The TPD method was used to test the desorption properties of NH3 and NO on the catalysts. The desorption properties measurement was carried out in a quartz tubular reactor (0.02 m in inner diameter), in which 7 mL of catalyst was placed. The catalysts were heated from room temperature to 500 ��C under a N2 atmosphere to eliminate any adsorbed water. After cooling to room temperature under the same atmosphere, NH3 (9��10-4, volume fraction) and N2 flowed to the catalyst. After 2 h, the physisorpted NH3 on the catalyst was purged with N2 flow until outlet NH3 could not be detected by FTIR. Finally, the samples were heated from room temperature to 500 ��C at a rate of 7 ��C/min under 1.44 L/min N2 flow, and the outlet NH3 was detected. The operation process of NO desorption was almost the same but using NO instead of NH3. The concentrations of NO, NO2, and NH3 were measured by gas analyzers using a Fourier transform infrared spectroscopy (FTIR) method (Bomen, MB154 model GASMET FTIR Dx4000). The Brunauer-Emmett-Teller (BET) surface area, pore volume, and average pore diameter of samples were measured by nitrogen adsorption porosimetry (Micromeritics ASAP-2020).
2.2 DRIFT experiment
In situ DRIFT was used to investigate the adsorption of NH3 and NO on the catalytic process. The DRIFT measurements were performed with VERTEX 70 FTIR spectrometers. The DRIFT spectra were recorded by accumulating 256 scans at a spectra resolution of 4 cm-1. In DRIFT cell, the catalyst was pretreated at 500 ��C in N2 environment for 2 h and then cooled to 180 ��C. The background spectrum was recorded in flowing N2 and subtracted from the sample spectrum. The spectra covering the range of 1 200-1 800 cm-1 were recorded to elucidate the chemical structure of NO and NH3 species adsorbed on catalysts.
2.3 Computational methods and model
For study of adsorption states with respect to transition-metal-oxide catalysts, the periodic boundary density functional theory is reliable, and can get rid of the restrictions of the cluster approach [13]. In this program, the one-electron Schr?dinger equations are solved only at the k=0 wave vector point of the Brillouin zone. Molecular orbitals were expanded into a set of numerical atomic orbitals of doubly numerical + polarization (DNP) quality provided by the software package. The DNP is comparable in quality to Pople��s split-valence 6-31G** basis set and usually yields the most reliable results [14]. To eliminate the convergence problems in some cases, partial occupancies were allowed for the highest orbitals using smear values of less than 0.02 eV.
The CuAl2O4 (100) surface is adopted to study the adsorption properties. The (100) surface consists of two CuAl2O4 formula units, and the lattice constants along both b and c are 8.064 ? (Fig.1(a)). The vacuum distance between neighbouring layers is set to be 9.80 ?. At such a distance, interaction between layers is tiny. To get a good compromise between accuracy and computational cost, the (100) surface is modeled by a single monolayer. The atomic coordinates of the adsorption site (Cu2+ ion) and the whole adsorbate molecule were optimized, whereas the positions of the other surface atoms remained unaltered from those of the fully optimized clean surface.
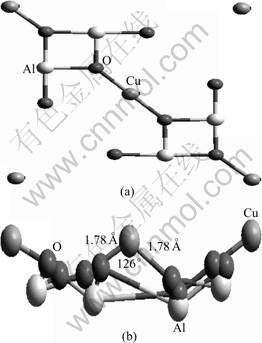
Fig.1 View of CuAl2O4(100) surface: (a) Top view of CuAl2O4 (100) surface; (b) Side view of fully relaxed CuAl2O4(100) surface
As already described, the (100) surface slab with Cu2+ ions is created with b=c=8.064 ?, where two formula units of CuAl2O4 are contained (Fig.1(a)). The (100) surface is fully optimized where the calculated b and c are 7.694 and 7.687 ?, respectively. The equilibrium bond length of Cu��O is 1.78 ?, and the corresponding O��Cu��O bond angle is 126.1��, as shown in Fig.1(b). Then, free molecules of NH3 and NO are calculated using a periodic boundary cubic cell with a cell length of 8 ?, and the structural parameters obtained, including the bond length and the bond angle, are listed in Table 1. It shows that the calculated values are in good agreement with experiment data [14].
Table 1 Calculated bond lengths and/or bond angles of free adsorbate molecules

3 Results and discussion
3.1 Catalyst characterization and catalytic activity on CuO/��-Al2O3
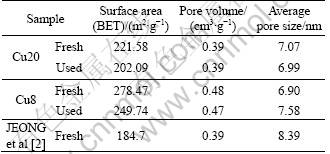
The BET surface area, pore volume, and pore size of Cu8 and Cu20 are summarized in Table 2. As shown in Table 2, the CuO/��-Al2O3 catalysts made by the sol-gel method have a larger surface area and pore volume than those made in the wet-impregnation method [2-3]. The excellent pore structure favors pore diffusion of the reactant gas on the catalyst surface [2], although the surface area is not a determining factor for activity. The surface area and pore volume decrease slightly after 100 h SCR test, which indicates that the catalysts have a strong capability of anti-ablation. The samples can be recycled.
Table 2 Pore structural parameters of fresh and used catalysts
The variations of the NO conversion as a function of temperature are shown in Fig.2. For CuO/��-Al2O3 catalysts, the activity is improved with the temperature increasing. The SCR activity increases with higher Cu loading. Clearly, high catalytic activities are shown in a lower temperature region (100-300 ��C). The NO conversion is nearly 100% at 300-400 ��C. The SCR activity decreases gradually when the temperature is higher than 400 ��C.
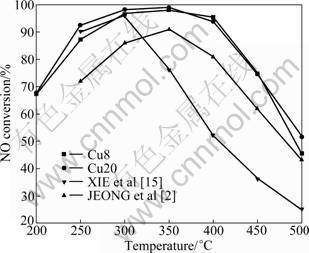
Fig.2 Effect of reaction temperature on NO conversion over CuO/��-Al2O3 catalyst (Reaction conditions: ��(NO)=7.5��10-4; ��(NH3)=9��10-4; ��(O2)=5%; vGHSV =12 800 h-1)
The catalysts of JEONG et al [2] and XIE et al [15] were prepared by wet-impregnation method. The active component contents of them are both 8% (mass fraction), and they are named as JEONG and XIE, respectively. Compared with the results of JEONG and XIE, the CuO/��-Al2O3 catalysts synthesized by the sol-gel method have higher activity over the whole temperature range. Especially, the Cu8 and Cu20 catalysts maintain 75% activity even at 400 ��C.
3.2 Desorption of NH3 and NO on CuO/��-Al2O3
The SCR reaction belongs to the gas-solid multiphase reaction, and the adsorption of NH3 and NO plays an important role in the reaction. The TPD method was used to test the desorption properties of NH3 and NO on the catalysts.
3.2.1 Desorption properties of NH3
The process of the NH3 desorption as a function of temperature is shown in Fig.3. There is an obvious NH3 desorption peak at 150-200 ��C on Cu8. The desorption peak of NH3 on Cu20 is un-conspicuous. It can be seen that the NH3 could be adsorbed on the catalysts and then desorbed when the temperature increases. The adsorption of NH3 on Cu8 is much stronger than that on Cu20. This difference between Cu8 and Cu20 may be due to the increase of the basicity caused by the Cu2+ on the catalysts. The basicity on the catalyst decreases the NH3 adsorption capacity, but the adsorption capacity at the low temperature (200-350 ��C) is not the key factor of the catalytic activity.
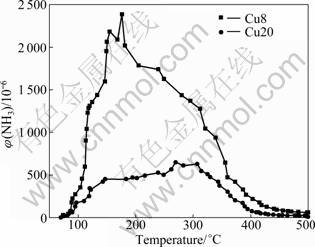
Fig.3 NH3 desorption as function of operating temperature over CuO/��-Al2O3 catalyst
In low temperature range (200-300 ��C), the adsorbing amount of NH3 on Cu8 is larger than that on Cu20. Compared with the catalytic activity in Fig.2, Cu8 has lower activity instead. It could be inferred that the adsorption capacity at low temperature is not the key factor of the catalytic activity. With the temperature increasing, especially at the high temperature (450- 500��C), the activity of Cu20 declines more sharply than Cu8. This might be related with the weak adsorption and the strong oxidation property of Cu20.
3.2.2 Desorption properties of NO
The variations of the NO desorption, and NO2 formed during reaction as a function of temperature are illustrated in Figs.4(a), and (b), respectively. There are two peaks of NO desorption at 100-150 ��C and 400- 450 ��C on Cu20. The desorption peaks of Cu8 is un-conspicuous. These desorption peaks infer that NO adsorbed on the catalysts exists in several forms. Most of adsorbed NO on the catalysts has been desorbed when the temperature is higher than 475 ��C.
NO2 is detected in the NO desorption process. There are two NO2 desorption peaks at 150-250 ��C and 350-450 ��C on Cu20. It is inferred that adsorbed NO is oxidized to form nitrate-like species. The desorption peak of NO2 on Cu8 is also un-conspicuous. In general, the desorption amount of NO and NO2 on Cu20 is larger than that on Cu8. The Cu2+ on the catalyst promotes the adsorption capacity of NO.
The desorption processes of two samples are almost the same; the desorption amount of NO on Cu20 is larger than that with Cu8. However, the desorption capacity of NH3 on Cu20 is smaller than that on Cu8 in Fig.3. As a conclusion of this work, it is proposed that the adsorption mechanism of NH3 and NO on the two samples is different and NO is more strongly adsorbed than NH3.
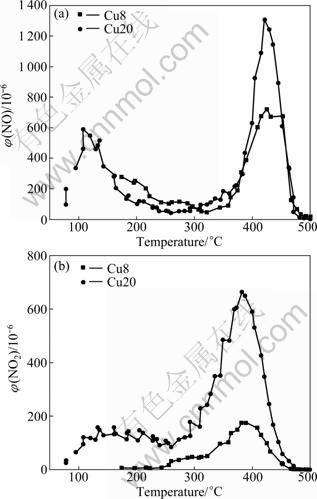
Fig.4 NO desorption as function of operating temperature over CuO/��-Al2O3 catalyst: (a) NO desorption; (b) Volume fraction of NO2
3.3 Reaction kinetics of CuO/��-Al2O3
Following the Eley-Rideal mechanism, the reaction rate is proportional to the gas-phase concentration of NO and the fraction of the surface covered by the adsorbed NH3. The reaction rate is reduced to the first order for NO and zero order for NH3 when NH3 is strongly adsorbed on catalysts. Therefore, the intrinsic reduction rate of NO can be expressed as
r(NO)=Kr��c(NO) (1)
where r(NO) is the intrinsic reaction amount of NO, mol/(g��s); c(NO) is the concentration of NO at the entrance of the catalyst bed, mol/L; Kr is the intrinsic reaction rate, mol/(g��s).
Kr can be calculated with an assumption of the gas phase flows in plug flow as
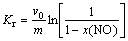 (2)
(2)
Also,
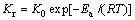 (3)
(3)
where v0 is the total volumetric flow rate of the gas phase at the reactor entrance, m3/s; m is the mass, g; x(NO) is the conversion of NO; K0 is the frequency factor of the intrinsic rate constant; T is the reaction temperature, K; Ea is the activation energy of the reaction.
From Arrhenius plots (Kr vs reaction temperature) for Cu8 and Cu20 (Fig.5), Ea and K0 of the catalysts are derived. The pre-exponential factor and activation energy of the catalysts are listed in Table 3.
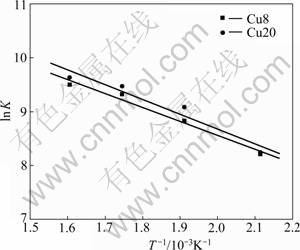
Fig.5 Arrhenius plots of intrinsic reaction rate constants for CuO /��-Al2O3
Table 3 Activation energy and pre-exponential factor of catalysts
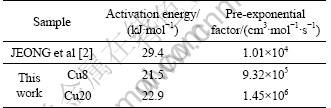
The kinetic parameters of JEONG et al [2] are also included in Table 3. It can be seen that the the activation energy of Cu8 and Cu20 is lower than that of JEONG et al. It is consistent with the high activity of Cu8 and Cu20 at the temperature ranges. This may be related with the pore property of the catalysts. As shown in Table 2, the CuO/Al2O3 catalysts made by sol-gel method have larger surface area and pore volume than those made by wet-impregnation method. The excellent pore structure favors pore diffusion of the reactant gas on the catalyst surface.
3.4 DRIFT study of NH3 and NO on CuO/��-Al2O3
3.4.1 Adsorption peaks of NH3
Figure 6 shows the DRIFT spectra produced from the co-adsorption of NH3 at 180 ��C and 300 ��C on Cu20. Flowing NH3+O2 at 180 ��C produces the bands at 1 620, 1 598, 1 580, 1 460, 1 395, 1 375, 1 250 and 1 230 cm-1. The bands at 1 620, 1 598, 1 580, 1 395, 1 375, 1 250- 1 120 cm-1 could be attributed to co-ordinate ammonia species on Lewis acid site [5-6, 16-17]. The bands at 1 460 cm-1 could be assigned to ammonium ions on Br?nsted acid sites [6]. Increasing the temperature to 300 ��C causes the 1 250 cm-1 band to sharpen, the 1 620, 1 230 cm-1 band to disappear gradually and the intensity of 1 395 and 1 375 cm-1 band to decrease.
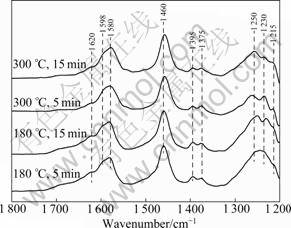
Fig.6 DRIFT spectra of CuO/��-Al2O3 exposed to NH3 for various times at 180 ��C and 300 ��C
RAMIS et al [16] investigated the SCR mechanism over CuO/TiO2 at 200-250 ��C by FTIR. They found that the NH3 adsorbed on the Lewis acid sites was activated through H-abstraction (partial oxidation) to form NH2 (amide), which then reacted with the gas phase NO to form a nitrosamide (NH2NO) species, an intermediate phase that was decomposed into N2 and H2O. This mechanism was termed amide-nitrosamide (NH2- NH2NO) mechanism. The formation of a band at 1 460 cm-1 assigned to HNO species [16] could provide evidence for the formation of N��O bonds from ammonia. The formation of NO and N2O during the NH3 oxidation process also confirmed the amide�Cnitrosamide mechanism.
3.4.2 Adsorption peaks of NO
Figure 7 shows the DRIFT spectra produced from the adsorption of NO at 180, 250 and 350 ��C on Cu20. Flowing NO at 180 ��C produces the bands at 1 620, 1 585, 1 560, 1 395, 1 298 and 1 244 cm-1. The bridging bidentate nitrate at 1 620 cm-1 is due to the dispropor- tionation of NO [5, 18-19]. The bands at 1 244 cm-1 could be attributed to chelating nitro. The bands at 1 298, 1 585 and 1 560 cm-1 are assigned to chelating bidentate nitrate [18] .
The intensity of bands at 1 200-1 640 cm-1 describes the amount of nitrate on the catalyst surface. Increasing the temperature to 300 ��C causes the intensity of the bands at 1 560 cm-1 to increase, which suggests that adsorbed nitro species are oxidized to nitrate species. High temperature is responsible for the low amount of NO adsorbed on samples. It can be seen that NO is difficult to adsorb on the catalyst at high temperature. The NO desorption process (Fig.4) also confirms that NO and NO2 are desorbed with the temperature increasing.
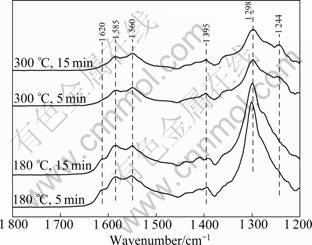
Fig.7 DRIFT spectra of CuO/��-Al2O3 exposed to NO for various times at 180 ��C and 300 ��C
3.5 DFT study of NH3 and NO on CuAl2O4 (100) surface
3.5.1 Adsorption states of NH3
When an NH3 molecule approaches the Cu2+ site, adsorption takes place. The adsorption energy is calculated to be -570.36 kJ/mol, indicating a strong interaction between the adsorbate and substrate. As demonstrated in Fig.8 and Table 4, the N��Cu bond is found to be 1.93 ?, perpendicular to the (100) plane. With NH3 adsorption, the Cu��O bond is elongated by 0.06 ?, and the O��Cu��O angle changes from 126.1�� to 119.6��. In general, when the adsorption occurs, the bonding ability in the adsorbate species might be weakened; accordingly, the atomic distance might be elongated. This is expected to happen in the case of NH3 adsorption. Surprisingly, the N��H bond lengths almost remain unchanged. There might be a shortening of the N��H bonds, and such a shortening may be offset by the elongation resulted from the adsorption. When turning to electronic properties, the charges on the N and H atoms are larger than the corresponding charges in free NH3 (Table 5). The increased atomic charge in the adsorbate species implies that the interaction between the N and H atoms increases correspondingly. As a result, the N��H bond length should be decreased. As expected, this contraction offsets the N��H bond expansion and the net result shows that the N��H bond length is nearly unaltered.
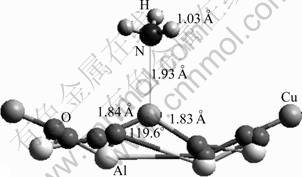
Fig.8 NH3 adsorption on Cu2+ site of CuAl2O4 (100) surface
On the other hand, it is observed that the adsorbed NH3 species is polarized, and has a charge of +0.177 (Table 5). This clearly indicates that the adsorbate donates 0.177 electrons to the substrate. As the adsorption site, as shown in Table 4, the Cu2+ decreases its charge by about 0.11, indicating that the Cu2+ receives about 65% of the electrons donated from the NH3 species.
3.5.2 Adsorption states of NO
Owing to the specific bonding property as described in previous study, either N or O atom of the NO molecule is able to interact with the Cu2+ active site and leads to different bonding configurations (Figs.9 and 10). When the N atom binds with the active site, the calculated adsorption energy of -610 kJ/mol shows that the adsorption is favorable energetically. The length of Cu��N bond formed is 1.78 ?, shorter than that of NH3. At the same time, the Cu��O bond is elongated by 0.08 ? and the O��Cu��O angle is decreased by 8.7��.
Regarding the change in the atomic charges, it is observed that the NO species becomes polarized with a net charge of 0.073 (Table 5). Obviously, the adsorption causes a charge flow and the NO donates 0.073 electrons to the substrate. Therefore, the substrate gets polarized as well. Most likely, the majority of the charge is expected to locate at the adsorption site. When concentrating on the charge of the Cu2+ ion, however, it is found that the charge on the site decreases by 0.200. This phenomenon shows that this site accumulates not only the charge of 0.073 transferred from the adsorbate, but also the charge of 0.127 (0.200-0.073) from its surrounding atoms within the substrate. This clearly demonstrates that the adsorption process leads not only to the charge transfer from the NO species to the adsorption site, but also to charge flow within the substrate. This process was also observed in previous adsorption studies.
Table 4 Selected structural parameters (bond lengths and bond angles) and atomic charges of adsorption site as well as adsorption energies with respect to adsorption of NH3 and NO on (100) surface (?q(Cu) represents change in charge of Cu2+ ion before and after adsorption)
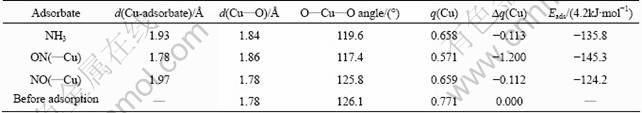
Table 5 Atomic charges of absorbates before and after adsorption, and net charges of adsorbate species due to adsorption
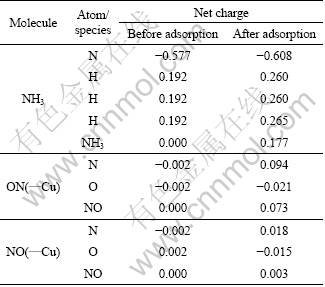
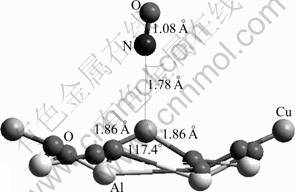
Fig.9 NO adsorption by interaction of N with Cu2+ on CuAl2O4 (100) surface
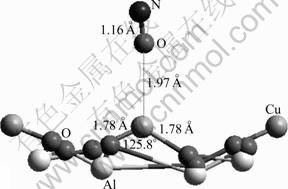
Fig.10 NO adsorption through interaction of O with Cu2+ on CuAl2O4 (100) surface
When the O atom of NO interacts with the active site, a stable adsorbate-substrate system is also obtained, and the interaction energy is calculated to be -521.64 kJ/mol (Table 4). This energy is the largest among all the cases treated in this work, which corresponds to the least stability. The newly formed Cu��O bond length is 1.97 ?, longer than that of the N��Cu bond for the NO adsorption by 0.19 ?. In the meantime, the changes of the Cu��O bond and the O��Cu��O angle are very little (Fig.10). This is also an indicator of the weak binding between the NO and the surface through the O and Cu interaction, and it is consistent with the calculated adsorption energy. Like the cases already studied, the adsorption site decreases its charge by 0.112 (Table 4), whereas the NO species is almost neutral with a net charge of +0.003 (Table 5). This indicates that the decrease of the charge on the Cu2+ site is mainly caused by the charge redistribution within the (100) surface.
3.5.3 Analysis of adsorption states
The adsorption energies demonstrate that all the adsorptions are stable energetically, and the stability decreases in the order ON(��Cu)>NH3>NO(��Cu) (Table 4). The adsorptions on CuAl2O4 surface are resulted from charge transfer between the adsorbate and the substrate, and the direction of the charge flow is from the adsorbate to the (100) surface. Eventually, both the adsorbate and the substrate get polarized. The adsorptions also lead to relaxation of the adsorption site, and the direction of the relaxation is outward from the surface.
The NO molecule has two different bonding forms to the Cu2+ with very different adsorption abilities as listed in Table 4. Therefore, only the one with the lower adsorption energy, ON(��Cu), is chosen to compare with the NH3 adsorption systems. Table 4 lists that the structural parameters and atomic charges on the active site are correlated well with the adsorption ability. With the decrease in the adsorption ability in the order ON(��Cu)>NH3, the Cu��O bond length in the substrate decreases accordingly, while the atomic distance between the Cu2+ and adsorbate, the O��Cu��O bond angle, the atomic charge and increased charge on the active site owing to the adsorption increase correspondingly. All these facts indicate that the change in the geometric and electronic structure induced by the adsorption of NH3 and NO is closely related to the adsorption ability.
NO adsorption energy is stronger than that of NH3. This phenomenon is consistent with the desorption ability of NH3 and NO in Figs.3 and 4. Compared with the desorption peak temperature of NO and NH3, it could be seen that peak temperature of NO is obviously higher than that of NH3. The stronger the adsorption capacity of NO on the Cu2+, the higher the temperature is needed to make chemical adsorption NO desorb form the catalysts.
From the DRIFT study of this work (Fig.6), it is known that NH3 can be strongly adsorbed on the catalysts, resulting in coordinated NH3 and NH4+. Figure 7 shows that NO adsorption leads to the formation of bridging bidentate nitrate, chelating bidentate nitrate, and chelating nitro. A high temperature is responsible for the low intensity of the NOx species. The adsorption peak intensity of NO is stronger than that of NH3 in Fig.4. This phenomenon is also in agreement with the results of the DFT analysis.
4 Conclusions
1) The CuO/��-Al2O3 catalysts made by the sol-gel method have higher activity and wider operating temperature range. The NO conversion of the catalysts maintains nearly 100% at 300-400 ��C.
2) There are obvious adsorption phenomena of NH3 and NO on the catalysts. NH3 can be strongly adsorbed on the catalysts, resulting in coordinated NH3 and NH4+. NO adsorption leads to the formation of bridging bidentate nitrate, chelating bidentate nitrate, and chelating nitro.
3) The adsorptions of all the molecules on the Cu2+ site of CuAl2O4 (100) are energetically favorable, whereas NO bound is the strongest with the adsorption site. The desorption peak temperature and adsorption peak intensity of NO both confirm this point.
4) The change in the geometric and electronic properties induced by the adsorption of NH3 and NO is correlated well with the adsorption ability. This work might be of help to understand the de-NOx mechanism and further to design de-NOx catalysts with excellent performance.
References
[1] CENTI G, PASSARINI N, PERATHONER S, RIVA A, STELLA G. Combined DeSOx/DeNOx reactions on a copper on alumina sorbent catalyst.3.DeNOx behavior as a function of the surface coverage with sulfate species [J]. Industrial and Engineering Chemistry Research, 1992, 31(8): 1963-1970.
[2] JEONG S M, JUNG S H, YOO K S, KIM S D. Selective catalytic reduction of NO by NH3 over a bulk sulfated CuO/��-Al2O3 catalyst [J]. Industrial and Engineering Chemistry Research, 1999, 38(6): 2210-2215.
[3] XIANG Jun, ZHAO Qing-sen, HU Song, SUN Lu-shi, SU Sheng, FU Peng, ZHANG An-chao, QIU Jian-rong, CHEN Han-ping, XU Ming-hou. Experimental research and characteristics analysis of alumina-supported copper oxide sorbent for flue gas desulfurization [J]. Asia-Pacific Journal of Chemical Engineering, 2007, 2(3): 182-189.
[4] ZHAO Qing-sen, XIANG Jun, SUN Lu-shi, SHI Jin-ming, SU Sheng, HU Song. Selective catalytic reduction of NO with NH3 over sol-gel-derived CuO-CeO2-MnOx/��-Al2O3 catalysts [J]. Journal of Central South University of Technology, 2009, 16(3): 0513-0519.
[5] RAMIS G, YI L, BUSCA G, TURCO M, KOTUR E, WILLEY R J. Adsorption, activation and oxidation of ammonia over SCR catalysts [J]. Journal of Catalysis, 1995, 157(2): 523-535.
[6] RAMIS G, LARRUBIA M A. An FT-IR study of the adsorption and oxidation of N-containing compounds over Fe2O3/Al2O3 catalyst [J]. Journal of Molecular Catalysis A: Chemical, 2004, 215(1/2): 161-167.
[7] LIU Qing-ya, LIU Zhen-yu, LI Cheng-yue. Adsorption and activation of NH3 during selective catalytic reduction of NO by NH3 [J]. Chinese Journal of Catalysis, 2006, 27(7): 636-646. (in Chinese)
[8] KIM T W, SONGA M W, KOHA H L, KIMA K L. Surface properties and reactivity of Cu/��-Al2O3 catalysts for NO reduction by C3H : Influences of calcination temperatures and additives [J]. Applied Catalysis A: General, 2001, 210(1/2): 35-44.
[9] LU GANGA, van GRONDELLEA J, ANDERSONA B G, van SANTENA R A. Selective low temperature NH3 oxidation to N2 on copper-based catalysts [J]. Journal of Catalysis, 1999, 186(1): 100- 109.
[10] SHIMIZU K I, MAESHIMA H, SATSUMA A, HATTORI T. Transition metal-aluminate catalysts for NO reduction by C3H6 [J]. Applied Catalysis B: Environmental, 1998, 18(1/2): 163-170.
[11] SHIMIZU K I, KAWABATA H, MAESHIMA H, SATSUMA A, HATTORI T. Intermediates in the selective reduction of NO by propene over Cu-Al2O3 catalysts: Transient in-situ FTIR study [J]. The Journal of Physical Chemistry B, 2000, 104(13): 2885-2893.
[12] LI Wei, CHENG Hua. Synthesis and characterization of Cu-Cr-O nanocomposites [J]. Journal of Central South University of Technology, 2007, 14(3): 291-295.
[13] YIN X L, FAHMI A, HAN H M, ENDOU A, AMMAL S S C, KUBO M, TERAISHI K, MIYAMOTO A. Adsorption of H2O on the V2O5(010) surface studied by periodic density functional calculations [J]. Journal of Physical Chemistry B, 1999, 103(16): 3218-3224.
[14] YIN X L, HAN H M, KUBO M, MIYAMOTO A. Adsorption of NH3, NO2 and NO on copper-aluminate catalyst: An ab initio density functional study [J]. Theoretical Chemistry Accounts, 2003, 109(4): 190-194.
[15] XIE Guo-yong, LIU Zhen-yu, ZHU Zhen-ping, LIU Qing-ya, GE Jun, HUANG Zhang-geng. Simultaneous removal of SO2 and NOx from flue gas using a CuO/Al2O3 catalyst sorbent: I. Deactivation of SCR activity by SO2 at low temperatures [J]. Journal of Catalysis, 2004, 224(1): 36-41.
[16] LARRUBIA A, RAMIS G, BUSCA G. An FT-IR study of the adsorption and oxidation of N-containing compounds over Fe2O3-TiO2 SCR catalysts [J]. Applied Catalysis B: Environmental, 2001, 30(1/2): 101-110.
[17] WU Zhong-biao, JIANG Bo-qiong, LIU Yue, WANG Hai-qiang, JIN Rui-ben. DRIFT study of manganese/titania-based catalysts for low-temperature selective catalytic reduction of NO with NH3 [J]. Environmental Science & Technology 2007, 41(16): 5812-5817.
[18] CHI Y W, STEVEN S C. Infrared study of NO adsorption and reduction with C3H6 in the presence of O2 over CuO/Al2O3 [J]. Journal of Catalysis, 2000, 190(1): 75-91.
[19] CENTI G, PERATHONER S. Nature of active species in copper-based catalysts and their chemistry of transformation of nitrogen oxides [J]. Applied Catalysis A: General 1995, 132(2): 179-259.
(Edited by YANG Bing)
Foundation item: Projects(50806025, 51021065, 50976038) supported by the National Natural Science Foundation of China; Project(20100480893) supported by the China Postdoctoral Science Foundation; Project(1001022B) supported by the Postdoctoral Research Fund of Jiangsu Province, China
Received date: 2010-06-29; Accepted date: 2011-05-03
Corresponding author: SUN Lu-shi, Professor; Tel: +86-27-87542417-8206; E-mail: sunlushi@mail.hust.edu.cn

