DOI: 10.11817/j.issn.1672-7207.2016.08.048
Fe2+���������������ȥ��ˮ�а�������
��ӱ1�������1��������2��������1
(1. �Ĵ���ѧ �����뻷��ѧԺ���Ĵ� �ɶ���610065��
2. �й�����������������о���Ժ����˾���Ĵ� �ɶ���610065)
ժҪ��Ϊ����Ч��ȥ��ˮ�е�Ũ�Ȱ��������ö�����(Fe2+)���������������(PMS)�������������գ���ˮ�а�����ȥ�������о����������첻ͬ��ʼpH������Ũ��(c(NH4+-N)0)��Fe2+��PMS���ʵ�����(n(Fe2+)/n(PMS))�������Ӽ���Fe2+/PMS��ϵȥ��ˮ�а�����Ӱ�졣�о��������������pH���ͣ�Fe2+/PMS��ϵ������ȥ��Ч����ǿ������n(Fe2+)/n(PMS)���Դٽ���ϵ������ȥ��������NH4+-N��ʼŨ�����ߣ�������ȥ���ʳ����½����ơ���pH=3��PMS��ʼͶ��Ϊ0.22 mmol/L��n(Fe2+)/n(PMS)Ϊ1:1��NH4+-N��ʼŨ��Ϊ0.044 mmol/L����Ӧ60 minʱ������ȥ���ʴﵽ���Ϊ88.27%����һ���棬�ֱ���Fe2+/PMS��ϵͶ�ӵ����ᡢ�����ᡢ����Ѫ��Ͳ���ȹ������Լ������Դٽ���ϵ������ȥ��Ч�������е��������ٽ�Ч����ѣ�ʹ����ȥ�������2.60%��Fe2+��Fe3+ת����Ч�ʼ��ߣ�ԼΪ91.8%��Fe2+/PMS����ȥ������������һ������ѧģ�͡�
�ؼ��ʣ�Fe2+�������������Σ���������������ɻ��������Ӽ�
��ͼ����ţ�X131.2 ���ױ�־�룺A ���±�ţ�1672-7207(2016)08-2900-07
Analysis on removal of ammonia nitrogen using peroxymonosulfate activated by Fe2+
YANG Ying1, GUO Hongguang1, DENG Qinzu2, ZHANG Yongli1
(1. College of Architecture & Environment, Sichuan University, Chengdu 610065, China;
2. Southwest Municipal Engineering Design & Research Institute of China, Chengdu 610065, China)
Abstract: In order to remove low concentration ammonia nitrogen in water effectively, remove the ammonia nitrogen in aqueous system by ferrous ion (Fe2+)/peroxymonosulfate (PMS), a new advanced oxidation process was analysed. The effects of initial pH, ammonia nitrogen concentration (c(NH4+-N)0), mole ratios of Fe2+ to PMS (n(Fe2+)/n(PMS)) as well as electronic agents on the removal of ammonia nitrogen were investigated. The results show that the removal rate is enhanced with the decrease of pH; the increase of n(Fe2+)/n(PMS) shows a positive effect, while the removal of contaminant exhibits a decline trend with the increase of initial NH4+-N concentration. At pH=3, 0.22 mmol/L PMS dosage, n(Fe2+)/n(PMS) at 1:1, and 0.044 mmol/L NH4+-N, the maximum removal rate of ammonia nitrogen is 88.27% after reacting for 60 min. On the other hand, adding tannic acid, citric acid, ascorbic acid and other electronic regent to this system can improve the efficiency of the removal rate. 2.60% increase of ammonia nitrogen removal rate is observed for the dosage of tannic acid. Meanwhile, the transformation of Fe2+ to Fe3+ is very high, which reaches 91.8%. The removal of ammonia nitrogen by Fe2+/PMS system fits the pseudo first order kinetics model.
Key words: Fe2+; peroxymonosulfate; ammonia nitrogen; sulfate radical; electronic agent
���������������������ˮ�帻Ӫ��������ˮˮ�ʲ�����¼����ֶ̬�ơ�ˮ�в���Ĺ�������������ˮ�����༰�������������ֳ��������ˮ�ܵ�����ˮ�豸�γ����ﹸ�������豸[1]��ˮ�����еİ�����Ҫ��Դ����ˮ��������ˮ��ũҵ������[2]�����������ˮ�������ܹ�ʹˮ��90%���ϵĺ����л������ȥ����������ĵ�����������ʽ�ŷţ��Ӷ���ʹ����Ӫ�����ʵ���Ⱦ��Ϊ��Ҫ�Ļ�������[3]����ͳ��ˮ�������նԹ���������ȥ�����ڵ�Ч�����ܵ�һ�����¶ȼ�Ӫ���������ƣ�Ϊ��ؽ�迪����Ч������ҵ��ˮ��ˮ�����а����������ա������ⳣ���İ���ȥ��������Ҫ��Ĥ���������ﻯ���������[2]�������ﻯ���еĸ��������վ��д������������١������ŵ���õ��㷺Ӧ�á�Ȼ��ǰ���о����ָù��ն�ԭˮ�Ľ�ˮˮ��Ҫ��ϸߣ��Ҵ����ɱ��߰�[4]��HUANG��[5]��ֻ����ǻ����ɻ�(��OH��E0=2.8 V)�Ĵ�ͳ�������������ܹ�����ȥ�����л���Ⱦ���ʣ�Ȼ������ˮ�а�����ȥ��ȴ��Ϊ��Ч(��OH��NH4+�ķ�Ӧ���ʽ�Ϊ9.7��107 L/(mol��s) [6])�����������о����ֹ��ɽ���(��Fe2+��Cu2+��Co2+ ��)�ܹ������������(S2O82-��PDS)�������������(HSO5-��PMS)������������ɻ�(��SO4-��E0=2.5~3.1V[7])(��ʽ(1)��(2))[8-11]������Ϊһ�������������գ��ܹ�����ȥ��ˮ�еİ���[12]��
 (1)
(1)
 (2)
(2)
���ڴˣ���������������ڡ�SO4-��������������ȥ�����ռ�Fe2+/PMS���գ������첻ͬpH��NH4+-N��ʼŨ��(c(NH4+-N)0)��Fe2+��PMS���ʵ�����(n(Fe2+)/n(PMS))�������Ӽ��ȶԸù��յĶ���ѧӰ�죬����Ϊ��ҵ��ˮ��ˮ�����й���������ȥ���ṩ�ο���
1 ����
1.1 ��Ҫ�Լ�
��Ҫ�Լ�Ϊ���Ȼ��������Ȼ�李����ᡢ�������ơ����������軯�ơ��������ơ������ᡢ����Ѫ�ᡢ���ᣬ��Ϊ�����������Գɶ�������ѧ�Լ�����˾������������ء��״�������Sigma-Aldrich��˾��������ˮ��Ϊȥ����ˮ��
1.2 ����������
1.2.1 ����Һ������
��ȡ1.91 g��100 ���������Ȼ��(NH4Cl)����ȥ����ˮ��(ȥ����ˮ�в����а�)�����Ƴ�500 L�İ���Һ������Һ����Ũ��Ϊ1.0 g/L(��N��)��
1.2.2 ���鷽��
������((25��2) ��)ȡһ������İ���Һ����500 mLƽ����ƿ�У�����0.1 mol/L��ϡ����(HNO3)����������(NaOH)������ҺpH���趨ֵ�������ִ���������ת��Ϊ160 r/min�����������趨��ӦҺ�м��벻ͬ�����Ȼ�����(FeCl2)ĸҺ(0.1 mol/L)�����������(PMS)ĸҺ(0.1 mol/L)��ͬʱ��ʼ��ʱ����Ӧ��ʼ�����趨ʱ��ڵ��϶�ʱȡ����Ѹ��Ͷ��0.1 mL�״�����ֹ��Ӧ(�״��롤OH��Ӧ����Ϊ9.7��108 L/(mol��s)���롤SO4-�ķ�Ӧ����Ϊ3.2��106 L/(mol��s)[13])����24 h�ڽ��ж���������
����ȥ����(��)�ļ��㹫ʽΪ
 (3)
(3)
ʽ�У� Ϊtʱ����Һ��NH4+-NŨ�ȵ�ʵ��ֵ��mmol/L��
Ϊtʱ����Һ��NH4+-NŨ�ȵ�ʵ��ֵ��mmol/L�� Ϊ��Ӧ��ʼʱ����Һ��NH4+-NŨ�ȵ�ʵ��ֵ��mmol/L��
Ϊ��Ӧ��ʼʱ����Һ��NH4+-NŨ�ȵ�ʵ��ֵ��mmol/L��
1.2.3 ��������
��Һ��NH4+-NŨ�Ȳ���ˮ����-�������α�ɫ���ⶨ[14]���ⶨ����Ϊ��UV-1800��MAPADA��������ֹ��ȼƣ���Ⲩ��Ϊ697 nm���������ݾ����豸��˾���������á�PinAAcle 900T����ԭ�������Dzⶨ��pH�����Ϻ��״š�pHB-4���ͷ����Dzⶨ�������Ϻ����ܿ�ѧ��������˾��
2 ��������
2.1 pH��Ӱ��
ǰ���о������������ϵpH�ĸı��ԡ�SO4-����������Ⱦ���������Ӱ������[15]��pH�Ա������Ӱ����Ҫ����������3�����棺һ����pH��Ӱ��Fe2+��NH4+��ˮ�еĴ�����ʽ���Ӷ�Ӱ��PMS�ļ���Ч����NH3�ij�ʼŨ�ȡ����⣬�ڲ�ͬpH�����£���ϵ�����ɻ����ͬ�����ڴˣ������鿼���ڲ�ͬpH������(3��5��7)��PMS��ʼͶ��Ϊ0.22 mmol/L��n(Fe2+)/n(PMS)Ϊ1:1��NH4+-N��ʼŨ��Ϊ0.044 mmol/Lʱ��Fe2+/PMS���ն�������ȥ��Ч���������ͼ1��ʾ��
��ͼ1��֪��Fe2+/PMS���ն�����ȥ����������pH����������͡���ʽ(4)��ʽ(5)��ʾ[16-19]��PMS��ˮ�в����ɲ�����SO4-������ͨ����ʽ��Ӧ�����ǻ����ɻ�(��OH)������ϵpHΪ����ʱ���ԡ�SO4-Ϊ��[16]�����������ȥ��ˮ�����������⣬����ʽ(6)[20]��ˮ�а�����Ҫ��NH4+��ʽ���ڣ�Ϊ�����ԣ������롤SO4-��ϣ��Ӷ������⡣����pH�������ߣ���SO4-����OHת���������ڡ�OH��������ȥ��ˮ�а������Ӷ�������ˮ�С�SO4-��Ũ�ȡ�
 ��
��
 (4)
(4)
 (5)
(5)
 (6)
(6)
���⣬����ʽ(7)~(9)[20-21]��pH��������ʹFe3+��Fe2+���������������ת����������Fe2+��PMS�ļ���Ч�ܣ��Ӷ������˰�����ȥ���ʡ�ZOU��[22]��Fe2+����PMS���о���Ҳ���֣���pH=3ʱ��Fe2+/PMS��ϵ������������ǿ��
 ��
��
 (7)
(7)

 (8)
(8)
 (9)
(9)

ͼ1 ��ͬ��ʼpH�°�����ȥ��
Fig. 1 Removal of ammonia nitrogen at different initial pH values
2.2 n(Fe2+)/n(PMS)��Ӱ��
��Fe2+/PMS��ϵ�У�Fe2+��Ϊ���������ɼ���PMS(E0=1.85 V)���������Ը�ǿ�ġ�SO4-(E0=2.5~3.1 V)�����Ҳ�ͬ��Fe2+Ͷ����Ӱ�졤SO4-�IJ���[23]�����鿼������pH=3��PMS��ʼͶ��Ϊ0.22 mmol/L��NH4+-N��ʼŨ��Ϊ0.044 mmol/Lʱ��ͨ���ı���ϵ��Fe2+��Ͷ����̽��Fe2+/PMS��Ͷ���ȶ���ȥ���ʵ�Ӱ�졣
��ͼ2��֪������Fe2+Ͷ���IJ�����������ȥ��Ч��Խ��Խ�á�����ʽ(4)��(6)��(10)������Fe2+Ͷ���IJ���������PMS������������SO4-������������NH4+���ϱ�ת��ΪNO2-��NO3-��N2������[24]������ȥ������n(Fe2+)/n(PMS)Ϊ1:1ʱ�ﵽ���ԼΪ88.27%��

ͼ2 pH=3ʱ��ͬn(Fe2+)/n(PMS)�°�����ȥ��
Fig. 2 Removal of ammonia nitrogen at different n(Fe2+)/n(PMS) at pH=3
 [6-8] (10)
[6-8] (10)
 ��
��
 [25-26] (11)
[25-26] (11)


 [27] (12)
[27] (12)

 [27] (13)
[27] (13)
 [8] (14)
[8] (14)
����n(Fe2+)/n(PMS)������2:1ʱ��������ȥ���ʷ������͡�����������ʽ(10)~(14)��ʾ��������Fe2+�ᱻת��ΪFe3+��Fe3+���ܿɼ���PMS����ȥ��ˮ�а�����������ANIPSITAKIS��[8]���о�������Fe3+/PMS��ϵȥ����Ⱦ�����������Fe2+/PMS��ϵȥ����Ⱦ�����������ˣ���n(Fe2+)/n(PMS)Ϊ2:1ʱ��ˮ�а�����ȥ�����Եͣ�ԼΪ83.84%��
2.3 NH4+-N��ʼŨ�ȵ�Ӱ��
���鿼����Fe2+/PMS��ϵ�Բ�ͬ����Ũ�ȵ�ˮ���ȥ����������Ӧ��ϵpH=3��PMS��ʼͶ��Ϊ0.22 mmol/L��n(Fe2+)/n(PMS)Ϊ1:1������NH4+-N��ʼŨ��Ϊ0.044��0.088��0.110��0.220 mmol/L���ø�����ϵ������ȥ��Ч����ͼ3��ʾ��
��һ����Χ�ڣ�����ȥ��Ч������NH4+-N��ʼŨ�ȵļ�С��������ߡ�����ʽ(15)��NH4+-N��ʼŨ�ȼ�С��ʹ�á�SO4-�����Ŀ�������Խ��Խ�࣬���Կ��١���Ч��ȥ����������Ӧ40 min��ˮ�а�����Ũ�������ȶ�������NH4+-N��ʼŨ��Ϊ0.044 mmol/Lʱ�ﵽ��ͣ�ԼΪ0.005 mmol/L��WANG��[28]����Fe2+/PMS��ϵȥ��Ⱦ��ʱ����ָ���ϵ�Ե�Ũ��Ⱦ��ȥ��Ч���Ϻá�
 [20, 28-30] (15)
[20, 28-30] (15)

ͼ3 pH=3ʱ��ͬNH4+-N��ʼŨ���°�����ȥ��
Fig. 3 Removal of ammonia nitrogen for different NH4+-N initial concentrations at pH=3
2.4 �����ӻ�ԭ����Ӱ��
ǰ���о����������Fe2+/PMS������Fe3+��Fe2+��ת���Ǹù��յ����ٲ��裬�䴫�����������˸ù��ն���Ⱦ���ȥ��Ч������ʽ(7)~(14)��ʾ��ͨ������ϵͶ�ӹ����ӻ�ԭ��������һ���̶��ϴٽ�Fe3+��ԭ��Fe2+��������Fe3+���ۻ�[22]��������ͨ���ڷ�Ӧ�������������ϵ��Ͷ��0.22 mmol/L(��Fe2+�����ʵ�����Ϊ1:1)�ĵ����ᡢ�����ᡢ����Ѫ��Ͳ��ᣬ�ⶨ��Ӧ60 minʱ����ϵ������Ũ�ȣ��Կ���������ʶ�Fe2+/PMS����ȥ��������Ӱ�죬����ͨ����ⷢ�ָ���ϵ��Ͷ�ӻ�ԭ��ǰ��pH�������ֲ���(pH=3��0.4)��
��Ӧ60 minʱ����ͬ��ԭ���°�����ȥ������ͼ4��ʾ����ͼ4��֪����ͬ����Ļ�ԭ���Է�Ӧ�����ò�����ͬ���������3�ֹ��������ʣ����������ȥ���ʵ����Ч����������ԣ�ȥ����ԼΪ90.87%��ZOU��[22]�Ա�������ΪFe2+/PMS��������ϵ��Ŀ�����Ͷ���ǰ��Դٽ�Fe2+/Fe3+��ѭ������ʽ(16)��ʾ��ͨ����ԭ���ã��ǰ�����һ���̶�������Fe3+�ڸ���ϵ�е��ۻ����Ӷ��������������
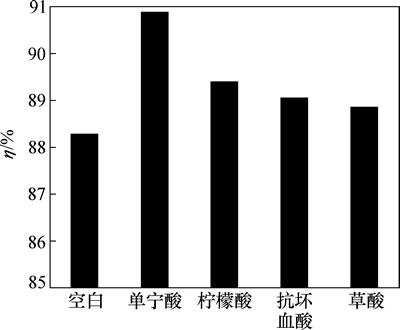
ͼ4 ��Ӧ60 minʱ��ͬ��ԭ���°�����ȥ����
Fig. 4 Removal of ammonia nitrogen at different reductants at 60 min
 (16)
(16)
���ǰ����ƣ�������Ⱦ���ǿ��ԭ���ұ��������е�Ԫ�أ��Ӷ��ɽ�Fe3+���ϵػ�ԭ��Fe2+�������ϵȥ��������������
2.5 ���ı仯���
��ʽ(10)��ʾ��Fe2+�ڼ���PMSȥ��ˮ�а����Ĺ����У������Fe3+ת�����������ԭ�������Ǻ�����ֹ����Dzⶨ��pH=3��PMS��ʼͶ��Ϊ0.22 mmol/L��n(Fe2+)/n(PMS)Ϊ1:1��NH4+-N��ʼŨ��Ϊ0.044 mmol/Lʱ����ϵ��Fe2+������Ũ�ȵı仯��
pH=3ʱFe2+��������Ũ���Լ�Fe2+��������Ũ�ȱȵı仯��ͼ5��ʾ����ͼ5�ɼ���Fe2+/PMS��ϵ�е���ʼ����������̬���ڣ���ˣ���Ӧǰ��������Ũ�ȼ������ֲ��䣬ԼΪ0.22 mmol/L����Ӧǰ20 min��Fe2+������Fe3+ת�����Ӷ�ʹFe2+�������еı�����1.00Ѹ�ٽ���Ϊ0.11���ҡ��ɴ˿�֪��Fe2+��PMS�ļ���Ч�����������ܲ��������ġ�SO4-ȥ����ˮ�а��������ʡ�Ȼ����Fe2+��Ũ�������40 min�ķ�Ӧ�����м����ޱ仯��˵��Fe3+�����ۻ�������Fe2+�Ľ�һ��ת�����ڸ����������£���Ӧ60 min��Fe2+��ת���ʴ�91.8%��
2.6 ����ѧ����
�����о�����Fe2+/PMS����ˮ������Ⱦ���ʷ�����һ������ѧģ����ϣ��������ೢ�Բ�����һ������ѧģ���������Ӱ�����ضԸù��ս���ˮ�а�����Ӱ�졣����ʽ(4)��(6)��(10)~(15)��ģ�Ϳɱ�ʾΪ
 (17)
(17)

ͼ5 pH=3ʱFe2+��������Ũ���Լ�Fe2+��������Ũ�ȱȵı仯���
Fig. 5 Changes of Fe2+, total iron concentrations and mole ratio of Fe2+ to total iron at pH=3
��ʽ(17)���л��ֲ�������
 (18)
(18)
�����ڲ�ͬ���������£�NH4+˥���İ�˥�ڶԱ������ȡc(NH4+-N)t=1/2c(NH4+-N)0��
 (19)
(19)
���У�kappΪ��һ���������ʳ�������ӳ��NH4+��Fe2+/PMS��ϵ���ڶ����ص�Ӱ������(���¶ȡ�pH��PMS��)��c(NH4+-N)0��c(NH4+-N)t�ֱ�ΪNH4+-N��ʼŨ�Ⱥͷ�Ӧtʱ�̵�Ũ�ȡ�������һ������ѧģ����ϲ�ͬ����·�Ӧ��ϵ����һ���������ʳ��������ϵ��r2����˥��t1/2��ϲ������1��ʾ��
ͼ6��ʾΪ��ͬ����������Fe2+/PMSȥ���������ʵ�Ӱ�졣�ɱ�1��ͼ6�ɼ���NH4+-N��ʼŨ�ȡ�n(Fe2+)/n(PMS)��pH��3��Ӱ�����ӷ������Թ�ʽ��NH4+-N��ʼŨ�����������ʽ��ͣ����������������(���ϵ��r2=0.92~0.99)��˵��NH4+������������Fe2+/PMS��ϵ��NH4+-N�Ľ������ʣ���n(Fe2+)/n(PMS)�����������������Ӻ��С�����ֵ��n(Fe2+)/n(PMS)Ϊ1:1����˵��Fe2+��PMS�����Եļ������ã�Fe2+Ũ�������ܴ�ʹPMS���ɸ�������������ɻ������NH4+-N�Ľ������ʣ�pH����ʹ�������ʼ�С�����������������(r2=0.93~0.97)��˵����������(pH=3)�¸���ϵȥ��NH4+-N��Ч���Ϻá�
��1 Fe2+/PMS��ϵ�а����ڲ�ͬ�����µ�ȥ����Ӧ����ѧ
Table 1 Kinetic of ammonia nitrogen removal at different conditions in Fe2+/PMS system


ͼ6 ��ͬ����������Fe2+/PMSȥ���������ʵ�Ӱ��
Fig. 6 Effects on removal rate of ammonia nitrogen at different conditions by Fe2+/PMS
3 ����
1) ��Һ��ʼpH������ȥ����Ӱ��ϴ�����pH=3ʱ��Fe2+/PMS��ϵ������ȥ��Ч����ѡ�
2) ��Fe2+/PMS��ϵ���ⰱ���ķ�Ӧ�У�n(Fe2+)/n(PMS)Ϊ1:1ʱ��Fe2+����PMS��Ч����ߡ�
3) Fe2+/PMS��ϵ�Ե�Ũ�ȵĺ�����Һȥ��Ч���Ϻã�������ϵpH=3��PMS��ʼͶ��Ϊ0.22 mmol/L��n(Fe2+)/n(PMS)Ϊ1:1��NH4+-N��ʼŨ��Ϊ0.044 mmol/Lʱ������ȥ������ߣ�Ϊ88.27%��
4) �����Ӽ���Ͷ�Ӷ�Fe2+/PMS��ϵ����Ӱ�첻���ԣ�Ͷ�ӵ��������ȥ���ʵ����Ч������������ᡢ����Ѫ��Ͳ����Ч�����Ϻã��ɴ�90.87%��
5) Fe2+/PMS��ϵ����NH4+-N��������pH��NH4+-N��ʼŨ�ȵ����Ӷ����ͣ�����n(Fe2+)/n(PMS)���������������С��������ϵpH=3��PMS��ʼͶ��Ϊ0.22 mmol/L��n(Fe2+)/n(PMS)Ϊ1:1��NH4+-N��ʼŨ��Ϊ0.044 mmol/Lʱ���ﵽ���
�ο����ף�
[1] KHUNTIA S, MAJUMDER S K, GHOSH P. Removal of ammonia from water by ozone microbubbles[J]. Industrial & Engineering Chemistry Research, 2013, 52(1): 318-326.
[2] HEGGEMANN M H, WARNECKE H J, VILJOEN H J. Removal of ammonia from aqueous systems in a semibatch reactor[J]. Industrial & Engineering Chemistry Research, 2001, 40(15): 3361-3368.
[3] HUANG Y, SONG C, LI L, et al. The mechanism and performance of zeolites for ammonia removal in the zeolite packed electrolysis reactor[J]. Electrochemistry, 2014, 82(7): 557-560.
[4] ��С��, ������, ������, ��. Fe2+�����������ط���������Ϣʹ[J]. ����������ѧѧ��(��Ȼ��ѧ��), 2015, 43(3): 137-142.
GUAN Xiaohong, XIN Xiaoyan, GAO Naiyun, et al. Degradation of acetaminophen by Fe2+ activated peroxymonosulfate oxidation[J]. Journal of South China University of Technology (Natural Science Edition), 2015, 43(3): 137-142.
[5] HUANG L, LI W, DONG Y, et al. Removal of ammonia by OH radical in aqueous phase[J]. Environmental Science & Technology, 2008, 42(21): 8070-8075.
[6] VON GUNTEN U. Ozonation of drinking water: part 1. oxidation kinetics and product formation[J]. Water Research, 2003, 37(7): 1443-1467.
[7] ������, �����, �˾�, ��. ���⼤��������ν���ˮ�п�����ƽ�о�[J]. ���пƼ���ѧѧ��(��Ȼ��ѧ��), 2013, 41(12): 117-122.
GAO Naiyun, HU Yuhao, DENG Jing, et al. Study on UV-activated persulfate oxidation of carbamazepine in water[J]. Journal of Huazhong University of Science and Technology (Nature Science Edition), 2013, 41(12): 117-122.
[8] ANIPSITAKIS G P, DIONYSIOU D D. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 2004, 38(13): 3705-3712.
[9] CHEN X Y, CHEN J W, QLAO X L. Performance of nano Co3O4/peroxymonosulfate system: kinetics and mechanism study using acid orange 7 as a model compound[J]. Applied Catalysis B: Environmental, 2008, 80(1/2): 116-121.
[10] HOUSE D A. Kinetics and mechanism of oxidations by peroxy disulfate[J]. Chemical Reviews, 1962, 62(3): 185-203.
[11] �˾�, ���Ʒ�, ������, ��. �������������θ����������о���չ[J]. ˮ��������, 2015, 41(4): 13-19.
DENG Jing, FENG Shanfang, MA Xiaoyan, et al. Research development in advanced oxidation processed based on homogeneous activation of peroxymonosulfate[J]. Technology of Water Treatment, 2015, 41(4): 13-19.
[12] ANIPSTTAKIS G P, STATHATOS E, DIONYSIOU D D. Heterogeneous activation of oxone using Co3O4[J]. The Journal of Physical Chemistry B, 2005, 109(27): 13052-13055.
[13] KEENAN C R, SEDLAK D L. Factors affecting the yield of oxidants from the reaction of manoparticulate zero-valent iron and oxygen[J]. Environmental Science & Technology, 2008, 42(4): 1262-1267.
[14] ���һ��������ܾ�.ˮ�ͷ�ˮ����������[M]. 4��. ����: �й�������ѧ������, 2002: 1-836.
State Environmental Protection Administration of China.Monitoring and analytic methods of water and wastewater[M]. 4th ed. Beijing: Environmental Science Press of China, 2002: 1-836.
[15] RASTOGI A, ALABED S R, DIONYSIOU D D. Sulfate radical-based ferrous�Cperoxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems[J]. Applied Catalysis B: Environmental, 2009, 85(3/4): 171-179.
[16] ABU AMR S S, AZIZ H A, ADLAN M N. Optimization of stabilized leachate treatment using ozone/persulfate in the advanced oxidation process[J]. Waste Management, 2013, 33(6): 1434-1441.
[17] BACHEL H W, PAUL G T, RICHARD L J, et al. Oxidation of chlorinated ethenes by heat-activated persulfate: kinetics and products[J]. Environmental Science ��Technology, 2007, 41(3): 1010-1015.
[18] �����, ������, ������, ��. �ȼ���������ν���ˮ�е��ͷ��ŵͪ�����ط���[J]. �Ĵ���ѧѧ��(���̿�ѧ��), 2015, 47(2): 191-197.
GUO Hongguang, GAO Naiyun, ZHANG Yongli, et al. Analysis on the degradation of typical fluoroquinolone in the water by thermally activated persulfate[J]. Journal of Sichuan University (Engineering Science Edition), 2015, 47(2): 191-197.
[19] ������, ����ƽ, ̸��Ⱥ, ��. �ȼ��������������������в�¡[J]. ����������ѧѧ��(��Ȼ��ѧ��), 2013, 41(12): 36-42.
GAO Naiyun, ZHU Yanping, TAN Chaoqun, et al. Degradation of diuron via heat-activated persulfate oxidation[J]. Journal of South China University of Technology (Natural Science Edition), 2013, 41(12): 36-42.
[20] LEE D K. Mechanism and kinetics of the catalytic oxidation of aqueous ammonia to molecular nitrogen[J]. Environmental Science & Technology, 2003, 37(24): 5745-5749.
[21] EVANS M G, GEORGE P U N. The Fe(OH)2+ and Fe(O2H)2+ complexes[J]. Trans Araday Soc, 1949, 45: 230-239.
[22] ZOU J, MA J, CHEN L, et al. Rapid acceleration of ferrous iron/ peroxymonosulfate oxidation of organic pollutants by promoting Fe(III)/Fe(II) cycle with hydroxylamine[J]. Environmental Science & Technology, 2013, 47(20): 11685-11691.
[23] WANG Z, BUSH R T, SULLIVAV L A, et al. Selective oxidation of arsenite by peroxymonosulfate with high utilization efficiency of oxidant[J]. Environmental Science & Technology, 2014, 48(7): 3978-3985.
[24] DENG Y, EZYSKE C M. Sulfate radical-advanced oxidation process (SR-AOP) for simultaneous removal of refractory organic contaminants and ammonia in landfill leachate[J]. Water Research, 2011, 45(18): 6189-6194.
[25] ANIPSITAKES G P, DIONYSIOU D D. Transition metal/UV-based advanced oxidation technologies for water decontamination[J]. Applied Catalysis B: Environmental, 2004, 54(3): 155-163.
[26] XU X R, LI X Z. Degradation of azo dye orange G in aqueous solutions by persulfate with ferrous ion[J]. Separation and Purification Technology, 2010, 72(1): 105-111.
[27] DE L J, LE T G. Kinetics and modeling of the Fe(III)/H2O2 system in the presence of sulfate in acidic aqueous solutions[J]. Environmental Science & Technology, 2005, 39(6): 1811-1818.
[28] WANG Y R, CHU W. Degradation of a xanthene dye by Fe(II)-mediated activation of oxone process[J]. J Hazard Mater, 2011, 186(2/3): 1455-1461.
[29] BALL R E, CHAKO A, EDWARDS J O, et al. Mechanism of oxidation of nitrogen nucleophiles by peroxodisulfate ion: nitrate ion and ammonia[J]. Inorganica Chimica Acta, 1985, 99(1): 49-58.
[30] LEE D K, CHO J S, YOON W L. Catalytic wet oxidation of ammonia: why is N2 formed preferentially against[J]. Chemosphere, 2005, 61(4): 573-578.
(�༭ ����ƽ)
�ո����ڣ�2016-01-08�������ڣ�2016-02-25
������Ŀ(Foundation item)���Ĵ�ʡ�����Ƽ��ƻ���Ŀ(2013HB08)��������Ȼ��ѧ����������Ŀ(51508354) (Project(2013HB08) supported by Sichuan Provincial Environmental Protection Office; Project(51508354) supported by the National Natural Science Foundation of China )
ͨ�����ߣ������������ڣ���ʿ����ʦ������ˮ���������о���E-mail: zxm581212@163.com

