���±��: 1004-0609(2005)01-0105-05
���ڷ��Ӷ���ѧ����������Au�����ܼ��㷽��
�� ΰ, �춨һ, ������, �����
(���ݴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ����350002)
ժ Ҫ: ����Ƕ��ԭ��ģ��, ���÷��Ӷ���ѧ���������˹����Au��ָ�����漰���ּ�ָ������ı����ܡ� ͬʱ, ����Levenberg-Marquardt �㷨, ������Au�����ܵ�BP������ģ��; ��Ϸ��Ӷ���ѧģ�͵ļ�������, ͨ���������ݵ���ѧϰѵ��, ���������ģ�Ͷ�Au��ָ����������ܵ�Ԥ�⡣ ����������: �÷������нϸߵ�Ԥ�⾫��, ����ȷԤ�Ե�ָ����������ܵ�����, Au������ı��������侧����(111)������нǵ����������������С���ص㡣
�ؼ���: ������; Ƕ��ԭ����; �˹�������; Levenberg-Marquardt�㷨 ��ͼ�����: TG146.31; TP183
���ױ�ʶ��: A
Calculation of Au surface energy by molecular dynamics combined with neural networks
TANG Wei, ZHU Ding-yi, CHEN Li-juan, GUAN Xiang-feng
(School of Materials Science and Engineering, Fuzhou University, Fuzhou 350002, China)
Abstract: Via embedded-atom model and molecular dynamics simulation, the surface energies of three low-index and some high-index planes were calculated for precious metal Au, and the error back-propagation network (BP) developed by Levenberg-Marquardt algorithm was adopted. Combining the data calculated with the molecular dynamics model, a great deal of data were trained many times and compared with the calculated data, and the prediction of high-index surface energy was performed. The results show that the method has high predicting accuracy. The order of the three low-index planes was predicted exactly. The surface energies on the other planes show a tendency that first increasing and then decreasing with angle between the planes and (111) plane increasing.
Key words: surface energy; embedded-atom potential; artificial neural network; Levenberg-Marquardt algorithm
�������DZ������ϱ������Ե���Ҫ����, �ڱ��桢 ǿ������ѡ� ���Ⱥܶ����������о����й㷺Ӧ�á� ���ھ����ڽṹ�ϵĸ�������, ��ͬ�ľ��������в�ͬ�ı�����, ����������Ҳ�кܴ���졣 Au�ڵ��ӹ�ҵ�ͼ�˿�ѧ���������Ǽ�����Ҫ�Ĺ��ܲ���, �ڵ��ӹ�ҵ��90%���ڱ���Ʋ㡣 �ƽ����߲��ϵ���ʴ��, �ƽ����һ�������ǶԺ�����ǿ�ҵķ�������, �㷺���ں���̽���Ƿ�����װ����, Au�ѳ�Ϊ�ִ�����ͨѶ�����еĹؼ�����[1]�� ������, Au�ı�������õõ��㷺��Ӧ��[2]�� ����о�Au�ı����ܾ�����Ҫ���塣 Ŀǰ����ͨ��ʵ�龫ȷ�ⶨ��̬���ϵı�����, ��ʹͨ��ʵ���ܻ��һЩ�ṹ����ı�����, �����һ�㶼�Ƚϴ� �������ܹ��ɵ�һԭ�������ڳ����������ֱ�Ӽ���һЩ�ṹ����ı�����, ����Ҫ���Ѵ�����ʱ, ͬʱҲ����һ�������[3]�� ����ͨ���÷�������ʵ�������¸��ָ��ӽṹ����ı�����, Ŀǰ������������ٶȺʹ洢������ԶԶ������ �����������, ���ý�����Ѹ�ٷ�չ�ĸ��ֶ�������, ����Ϸ��Ӷ���ѧ�������м����ģ�������ϵı�����, ������Ŀǰȷ����̬���ϱ����ܵ���Ч����, ������һ����ȷ��, �ֶԼ����Ҫ��̫��, �������߲���Ƕ��ԭ��ģ�ͶԹ����Au�ĵ�ָ�������ָ�ָ������ı����ܽ��з��Ӷ���ѧģ�����, �����ڸ��Ӹ�ָ������������÷��Ӷ���ѧ������������, Ŀǰ���б���, ��������̽���Եز����˹������緽��, ��Ϸ��Ӷ���ѧģ��ļ�������, ͨ���������ݵ���ѧϰѵ��, ���������ģ�ͶԸ�ָ����������ܵ�Ԥ��, ����ʵ�������бȽϡ�
1 Ƕ��ԭ��ģ�ͼ�������ȷ��
EAM����[4]�����ܶȷ������ۻ�����,������Ч���ʻ�ԭ�ӽ����Ƶ�������,������������Ҫ�ļ���:һ��ԭ�ӵ����ܶȷֲ�������ƽ������;���ǻ�������ܶ���ԭ�ӵĵ����ܶȵ����Ե��ӡ� ����EAM����,һ��ԭ�Ӽ��ŵ��������ɱ�ʾΪ
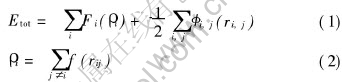
ʽ�� Fi(��i)ΪǶ����; ��i, j(ri, j)Ϊ������, ������Ҫ����ȡ��ͬ����ʽ; ��iΪ����i��ԭ���������������ԭ�ӵĺ�������ڵ�i��ԭ�Ӵ������ĵ������ܶ�֮��; f(rij)Ϊԭ�ӵ����ܶȷֲ�����; rijΪ��ԭ�Ӽ�ļ�ࡣ
Ҫ�õ�һ��Ƕ��ԭ��ģ��, ��ȷ��������������ʽ, ����Johnson[5]���ģ����㷽��:
F(��)=-Ec(1-lnx)x-6��ey(3)
f(r)=feexp[-��(r/re-1)](4)
��(r)=��eexp[-��(r/re-1)](5)
ʽ�� x=(��/��e)��/��; y=(��/��e)��/��; ��=3(��B/Ec)1/2; fe=Ec/��; ��e=Ec/6; �±�e��ʾƽ��״̬ʱ��ֵ��
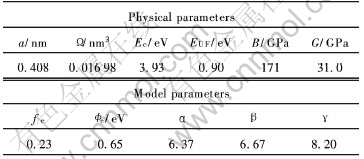
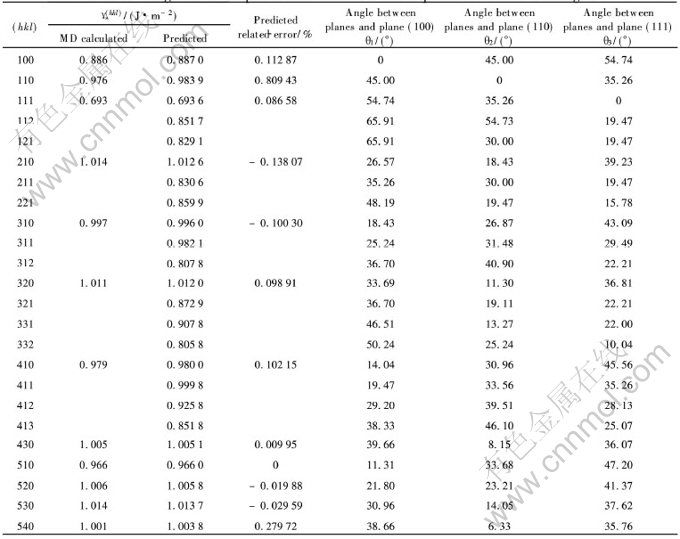
ͨ����Ͼ�����a�� ԭ��������� �ھ���Ec�� ����λ�γ���EUF�� ���ģ��B�ͼ���ģ��G�����Բ�����ͨ����ϻ�õ�ģ�Ͳ������ڱ�1��
2 �˹�������ԭ����ģ��
2.1 �˹�������ԭ��
��1 �����Au������������ģ�Ͳ���[5]
Table 1 Physical parameters and model parameters of precious metal Au
�˹�������(ANN)��Ŀǰ������Ѹ�ٷ�չ��ǰ�ؽ���ѧ��, �䱾���ǽ���һ��ӳ���ϵ, �ڹ���Ӧ�������ڵ��͵������ھ����� �˹�����������������Ϊ����, ����ij���㷨�������ε������һ����ӳ�����������ڹ��ɵ���ѧģ��, ��˾����ʺ����о����ӷ�����ϵͳ�Ͳ�ȷ�����̵��ŵ�, ���й㷺��Ӧ��ǰ��[6-8]�� ��������������(BP����)���н�ǿ���������ͷ�������, ����ѡ��BP���硣 BP����ͨ�������������������������������, ������������Ȩֵ����ֵ,ֱ������ﵽ��ƽ���Ҫ�����ӦΪֹ�� ����һ������㡢 һ�������㡢 һ��������3��BP��������ͼ1��ʾ(ͼ��, P������������; w1�� w2�ֱ�����������������Ȩֵ����; b1�� b2�ֱ�����������������ƫ������(��ֵ); a1�� a2�ֱ������������������������; n1�� n2�ֱ������������������Ԫ���������)�� ����BP�㷨�Dz��������½���̬Ѱ�ŵ�һ���㷨,����Ŀ�꺯���ļ�ֵ�㸽�������ٶ���, ����������㷨(���˹-ţ�ٷ�)��Զ��Ŀ�꺯���ļ�ֵ��ʱ�����ٶȺ���[8]�� ���о�����һ��������㷨����Levenberg-Marquardt�㷨[9], �����������ʽΪ
��w=(JTJ+��J)-1JTe(6)
ʽ�� JΪ������Ȩֵ�����ֵ�Jacobian����; eΪ�������, ����ij�����Խ�������Ӧ�����ķǸ����� ���������������ڦ̵ķ�ֵ�ĸı�, �⻬�������ּ���֮��仯: �����ĸ�˹-ţ�ٷ�(�� ��0)�;�����ݶȷ�(�̡���), ��ַ����������㷨������[10]��
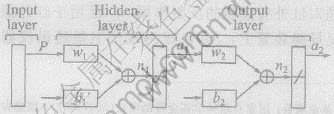
ͼ1 ����BP������
Fig.1 Three layers BP neural network
2.2 ������Ԥ��������ģ��
2.2.1 BP������ģ�͵Ľ���
����������3����ָ������((100)�� (110)��(111))�ļн���Ϊ������ѵ��������ʸ��, ����������ϵ��������(h1k1l1)��(h2k2l2)��ļнǹ�ʽ:
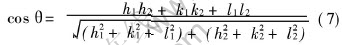
����ø�������(100)�� (110)��(111)3����ָ������ļн�, ������Ӧ�ĸ��ݷ��Ӷ���ѧģ�����ı�����ֵ��Ϊ���ʸ��, �����������2�С�
����BP������ѡ��Sigmoid������Ϊ������Ԫ�Ĵ��ݺ����� ����S����������, Ϊ�˱�֤������Ԫ�ķ���������, ��ԭʼ����Ҫ���б�������, ����������������������ѧϰ�ٶȡ� Ϊ�˽�����������������, ʹ���ǵľ�ֵΪ0, ������Ϊ1, ���߶������������±�������:

ʽ�� xpΪ��������, ƽ��ֵ , ��
, ��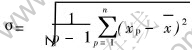 ��
��
Hecht-Nielson[11]֤���˶����κ��ڱ������ڵ�һ������������������һ���������BP�������ƽ�, һ�������BP���������������nά��mά��ӳ�䡣 ����ṹѡ�þ���һ���������BP����, ���������S�ͼ����, �����������Լ������
2.2.2 BP������Ԫ��Ŀ��ѡ��
��������Ԫ����Ŀ�Ǿ�����������Ч�Ժ�ȷ�ԵĹؼ�����֮һ, һ���������ĸ��ӳ̶Ⱥ�Ŀ��ľ���ѡ�����ʺϵ���Ԫ��Ŀ�� ���, ����Ҫ�趨������ڵ����������, ��ѵ��������Ĺ����в��ϵ���������ڵ���Ŀ�� ѵ��������ѵ�������, ֱ���ҵ����Ԥ������ ͨ�����ѵ���͵���, �ۺϿ��������ѧϰ�ٶȺͷ�������,����ȷ��6���ڵ��������, ��������˽ṹΪ3-6-1��
3 �����ܵļ���������
����������ģ����MatLab6.1�б��Ƴɿ����г���, ����������ļн�������Ϊ����ʸ��, ��������Ϊ���ʸ���ֱ��������ģ�ͽ���ѵ���� ��ѵ�����õ���Ȩֵ, �Ա����ܽ�����Ԥ��, ͬʱ��BP������Ԥ��������ֵ�����2�С� �ɱ�2��֪, ��ѵ��������Ա�����Ԥ�����Χ�DZȽ��ȶ��ͺ����ġ� ͼ2��ʾΪ�����ܵ�������Ԥ������MD�������ıȽ�, ������Ԥ������ܺͼ�������ܵ�����������ߡ� ��ͼ���Կ���, Ԥ��ֵ��MD����ֵ�нϸߵ����������, �������ϵ��R=0.99965�� �ɴ˿�֪, ��Ԥ��ģ�;��иߵ�Ԥ�⾫�ȡ� ƽ���������[TX-]���������¹�ʽ����:
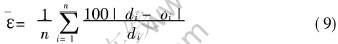
ʽ�� diΪĿ�����ֵ; oiΪԤ�����ֵ, nΪ���������Ŀ�� ������ɵ�: 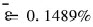 , ���������ܵļ���ֵ��Ԥ��ֵ�dz��ӽ�, ��˵�����潨����ģ�;��нϸߵľ��ȡ� ��Ҫָ������, ���ڱ������߶����ݽ����˱�������, ���������Ƶ�Ŀ������ǶԹ�һ��������ݶ���, �����������������Ƕ�ԭʼ���ݶ��Եġ� ����һ��������, �ؼ���������ṹ�� Ȩֵ����ֵ�� һ������ѵ�����, ��Щ���ݾͿ��Եõ�, �����DZ�������, ��д��Ԥ�⺯��, �ٴν���ѵ��, �������ٶ����������ѵ��, Ԥ����ٶȴ��ӿ졣
, ���������ܵļ���ֵ��Ԥ��ֵ�dz��ӽ�, ��˵�����潨����ģ�;��нϸߵľ��ȡ� ��Ҫָ������, ���ڱ������߶����ݽ����˱�������, ���������Ƶ�Ŀ������ǶԹ�һ��������ݶ���, �����������������Ƕ�ԭʼ���ݶ��Եġ� ����һ��������, �ؼ���������ṹ�� Ȩֵ����ֵ�� һ������ѵ�����, ��Щ���ݾͿ��Եõ�, �����DZ�������, ��д��Ԥ�⺯��, �ٴν���ѵ��, �������ٶ����������ѵ��, Ԥ����ٶȴ��ӿ졣
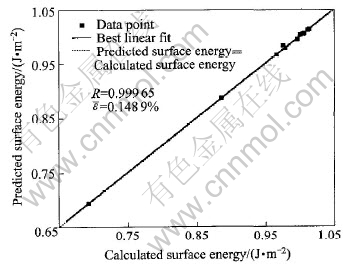
ͼ2 �����ܵ�������Ԥ������MD�������ıȽ�
Fig.2 Comparison of ANN-predicted and calculated surface energy
��������ģ��, ���߶Բ���δ֪����ľ����ܽ�����Ԥ��, �������ڱ�2�С�
����ѵ���������Ȩֵ������Է���, ������ʸ����3��ص�Ȩֵ���Ը���������������ʸ��, ����������ı����ܴ�С��Ҫ��������(111)�ļн��йء� �����ܦ����������(111)��нǦȵı仯��ͼ3��ʾ�� ���Կ���, ����������(111)������ı�������С, ��Ԥ�ڽ��һ�¡� ���������������������(111)��нǵ�����, �ڽǶȽ�Сʱ, �������Ե���������, ���ڦȡ�40�㸽��ʱ�ﵽ���ֵ, �ȼ�������, �����ܿ����½��� �Ա��ļ����õĸ����������ֵ�о�����, ������ֵ�����������½�������, ��ͬ���ڡ�001��������ĸ�����ָ������(��(410)��(510)��)�� ���ļ����õ�Au�����������������(111)����нǦȵı仯�Ĺ�ϵ���Ž����[12]���øĽ�Ƕ��ԭ�ӷ�����Cu����ı����ܵõ��ı��������ܶ����������(111)����нǦȵı仯���ƻ���һ�¡� ����Ԥ֪�����������������Ľ��������������������(111)����нǦȵı仯���ƻ����ϳ�����������С���ص㡣 �Ա�ͼ3�ͱ�2�����Է���, ����ͬ�����������н���ͬʱ, �ͱ��뿼��������������ָ������ļнǹ�ϵ��
��2 Au��ͬ������3����ָ������ļнǼ�������ı�����
Table 2 Angle between planes and three low-index planes and surface energies
������ɵ�, Ԥ������о�������ܵ�ƽ��ֵΪ0.9202J/m2, ͬʵ���õ�Au�ྦྷ��ı�����
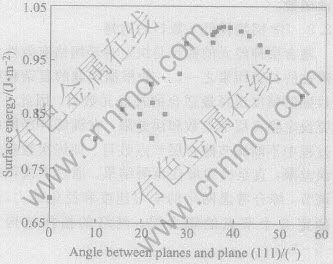
ͼ3 ���������������(111)����нǵı仯
Fig.3 Variation of surface energy with angle between planes and plane (111)
���ܶ�ƽ��ֵ1.50J/m2[13] �� Skriver�ĵ�һ��ԭ������ֵ[3]��Foiles��EAM����ֵ[14]�ȽϷ���, EAM����ģ�ͼ�������ܼ�������Ԥ��ı����ܾ����ڽ��ƫС�����⡣ ����Ҫ��: 1)ʵ��ⶨ������1.50J/m2�����о���ı����ܵ�ƽ��ֵ, �����ļ���ľ������; 2)��ͨ�����Ӷ���ѧ��������ļ�������û�п��ǵ�����ԭ�ӵ��ɳۡ� ��˽�����3�������������ʹԤ��������һ������ ��Skriver��һ��ԭ�������õ���������(111)����ı�����ֵΪ1.61J/m2, ��ʵ���õ�ƽ�������ܻ�Ҫ��, ͬ����ʾ��Skriver�����IJ��㡣 Ŀǰ��û��һ��ģ����㷽��������ȫ������㾧�������ͬʵ�����ݴ����������⡣ �ɱ�2�б�����Ԥ�����ݼ�ͼ2��֪������������ģ�Ϳ�����ȷԤ�Ե�ָ�����������ES������: ES(111)��ES(100)��ES(110), ������Ŀ��Ŷ�Խ��, �������Խ�� (111)����������, ����ı�������͡� �����, ����ԭ������Խ����, ������Խ�͡� ���ݹ����������ѧ���й�����[15], ����ԭ������Խ����ʱ, ԭ�Ӽ�������Խǿ, �Ӷ�ʹ���ϱ����ܽ���, �����������һ�µġ�
4 ����
1) ����������Ծ���нǺͱ�������������ѧϰ, ���øĽ���BP�����㷨����Levenberg-Marquardt�㷨�����˱������˹�������ģ��; ���������������ģ�;��нϸߵľ��Ⱥͺܺõķ��������� �÷�������ֱ���������������������������ܵļ���, Ҳ�������Ƶ������ľ���ṹ��
2) Au������������ı�����������(111)��нǵ�������ֳ���������С������, ���ڦȡ�40�㸽��ʱ�ﵽ���ֵ�� ������ֵ�����������½�������, ��ͬ���ڡ�001>������ĸ�����ָ������(��(410)��(510)��)��
3) ���Ӷ���ѧ����ı��������ݵ����徫�ȼ�������Ӱ�����ص�ȫ����, �����˶�Au����������ܵ�Ԥ�⾫�ȡ�
REFERENCES
[1]���ˮ, ����Ƽ. ��Ļ�ѧ[J]. �ߵȺ���ѧ��(��Ȼ��ѧ��), 2000, 13(1): 25-29.
RUAN De-shui, LI Wei-ping. Gold chemistry[J]. Journal of Higher Correspondence Education(Natural Science), 2000, 13(1): 25-29.
[2]���ذ�. �������еĽ�Ԫ��[J]. �����, 2003, 24 (1): 54-61.
DONG Shou-an.The role of gold in the nanotechnology[J]. Precious Metals, 2003, 24 (1): 54-61.
[3]Skriver H L, Rosengaard N M. Surface energy and work function of elemental metals[J]. Phys Rev B, 1992, 46(11):7157-7168.
[4]Daw M S, Baskes M I. Embedded-atom method: derivation and application to impurities, surface, and other defects in metals[J]. Phys Rev B, 1984, 29(12): 6443-6453.
[5]Johnson R A. Alloy models with the embedded-atom method[J]. Phys Rev B, 1989, 39(17): 12554-12559.
[6]QI Le-hua. Research on prediction of the processing parameters of liquid extrusion by BP network[J].Journal of Materials Processing Technology, 1999, 95: 232-237.
[7]Basheer L A. Artificial neural network: fundamentals, computing, design, and application[J]. Journal of Microbiological Methods, 2000, 43: 3-31.
[8]Joines J A, White M W. Improved generalization using robust cost functions[A]. IEEE/INNS Int Joint Conference of Neural Networks[C]. New York: IEEE Press, 1992. 911-918.
[9]Hagan M T, Menhaj M. Training feedforward networks with Marquardt algorithm[J]. IEEE Transaction on Neural Networks, 1994, 5(6): 989-993.
[10]������, �ﴺ��, ˮ��Ӣ��, ��. ��-ģ����������[M]. ����: ������ͨ��ѧ������, 1998. 156-215.
ZHANG Zhi-xing, SUN Chun-zai, Mizutani E, et al. Neuro-Fuzzy and Soft Computing[M]. Xi��an: Xi��an Jiaotong University Press,1998. 156-215.
[11]Hech-Nielsen R. Neurocomputing[M]. Massachusetts: Addison Wesley Publishing Company, 1991.
[12]�Ž���, ���Ϊ, ����. �øĽ�Ƕ��ԭ�ӷ�����Cu����ı�����[J]. ����ѧ��, 2003, 52(8): 1993-1999.
ZHANG Jian-min, XU Ke-wei, MA Fei. Calculation of suface energy of Cu crystal with modified embedded atom method[J]. Acta Physica Sinica, 2003, 52(6): 1993-1999.
[13]De Boer F R, Boom R, Mattens W C M, et al. Cohesion in metals[M]. Amsterdam, North-Holland: Elsevier Science, 1988. 716.
[14]Foiles S M, Baskes M I, Daw M S. Embedded atom method functions for the FCC metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys[J]. Phys Rev B, 1986, 33(12):7983-7990.
[15]����ʱ. ������Ͻ����о�����������[M]. ����: ��ѧ������, 1991. 21.
WEN Li-shi. Physics Basis of Solid Materials Interface Research[M]. Beijing: Science Press, 1991. 21.
������Ŀ: ������Ȼ��ѧ����������Ŀ(50471007); ����ʡ��Ȼ��ѧ����������Ŀ(A0210008)
�ո�����: 2004-05-25; ������: 2004-11-07
�����: �� ΰ(1981-), ��, ˶ʿ�о���.
ͨѶ����: �춨һ, ������; �绰: 0591-87893540; E-mail: zdy7081@163.com
(�༭�°���)

