文章编号:1004-0609(2007)09-1495-06
Zr5Si3及Zr3Ti2Si3弹性性质与热学性质
聂耀庄1, 2,谢佑卿2,彭红建2,李小波2
(1. 中南大学 物理科学与技术学院,长沙 410083;
2. 中南大学 材料科学与工程学院,长沙 410083)
摘要:应用基于密度泛函理论的平面波赝势方法计算16H金属硅化物Zr5Si3及Zr3Ti2Si3的基态晶格参数,得出弹性常数、体弹性模量、弹性模量、剪切模量及泊松比等弹性性质。利用弹性常数计算德拜温度、格林奈森常数,并在德拜-格林奈森模型基础上计算这两种金属硅化物的各向异性热膨胀系数,由此得出Zr5Si3的a、c方向各向异性热膨胀系数(高温时)分别为8×10?6和15×10?6,对Zr3Ti2Si3(高温时)分别为11×10?6和13×10?6,与实验基本相符。根据方向体弹性模量、方向弹性模量及重叠布居数讨论两种材料各向异性热膨胀不同的原因。
关键词:第一原理计算;热学性质;弹性性质;金属硅化物
中图分类号:O 621.24 文献标识码:A
Elastic and thermal properties of Zr5Si3and Zr3Ti2Si3
NIE Yao-zhuang1, 2, XIE You-qing2, PENG Hong-jian2, LI Xiao-bo2
(1. School of Physics Science and Technology, Central South University, Changsha 410083, China;
2. School of Materials Science and Engineering, Central South university, Changsha 410083, China)
Abstract: A first-principles study on elastic and thermal properties of intermetallic silicides with 16H crystal structure, Zr5Si3 and Zr3Ti2Si3, was done, using the pseudopotential plane-wave method in the framework of the density-functional theory. The equilibrium lattice parameters of ground state at 0 K were calculated. The elastic constants, bulk modulus, elastic modulus, shear modulus, and Poisson’s ratio were obtained. Debye temperature and Grüneisen parameters were calculated from elastic constants. The anisotropic coefficients of thermal expansion (CTE) for a and c axes of Zr5Si3 and Zr3Ti2Si3 were calculated based on Debye-Grüneisen model. For Zr5Si3 (at high temperature), they are 8×10?6 and 15×10?6, respectively; for Zr3Ti2Si3 (at high temperature), they are 11×10?6 and 13×10?6, respectively. The results are in agreement with available experimental data. According to the calculated directional bulk modulus, directional elastic modulus and overlap population, the cause of different anisotropic CTE in these two systems was discussed.
Key words: first-principles calculations; thermal properties; elastic properties; intermetallic silicides
16H金属硅化物[1]因其优异的性能成为高温结构金属间化合物[2]的研究热点之一。由于它们大多具有较高的各向异性热膨胀[3],其应用受到限制。对于如何降低其热膨胀的各向异性,研究人员已提出了不少方案。如Zr3Ti2Si3的设计[4],使热膨胀的各向异性由较高的αa/αc≈2 (Ti5Si3和Zr5Si3)降为αc/αa≈1.2。Schneibei 等[5?7]报道了第三组元的添加对Mo5Si3与Ti5Si3热膨胀各向异性的影响。Fu等[8]计算了Mo5Si3的弹性常数,讨论了它的稳定性与成键特征。基于第一原理的热膨胀系数计算则由于工作量巨大,一直是具有挑战性的任务。完全基于第一原理的热膨胀计算主要限于立方晶系金属[9],对非立方晶系材料的计算仅见对于纤维锌矿ZnS[10]的研究报道。Fu等[11]在德拜?格林纳森模型的基础上计算了几种Mo-Si化合物的热膨胀系数,从弹性性质分析了它们各向异性热膨胀的起因,并从理论上讨论了添加Nb与V对Mo5Si3各向异性热膨胀[12]的影响。Hayashi等[13]则报道了添加Nb与W对Mo5Si3各向异性热膨胀影响的实验研究。上述研究对深入理解和揭示16H金属硅化物的物理特性、改善材料性能都具有重要意义。但对于Zr5Si3及Zr3Ti2Si3的弹性性质与热膨胀的理论研究还未见报道。另外,由于在许多时候弹性常数难以测量,因而弹性常数等物理量的理论计 算[14?15]也很有意义。本文作者用第一原理方法计算了Zr5Si3及Zr3Ti2Si3的电子结构与弹性性质,并在德拜?格林纳森模型的基础上计算了这两种金属硅化物各向异性热膨胀系数,得到的结论与已有实验数据基本符合,并对弹性性质与热膨胀各向异性的关系作了讨论。
1 理论模型与计算方法
16H金属硅化物有几种不同的晶体结构。Zr5Si3及Zr3Ti2Si3具有D88(六方)结构,如图1所示。3个基本原子面按ABACA的顺序沿c轴方向堆垛,原子面A仅含金属原子,而原子面B与C含有金属原子与硅原子。B与C两个原子面的原子排列相当于绕中心轴旋转了180?。硅原子在原子面B与C上占据6g位置,原子面A上的金属原子占据4d位置。对Zr5Si3,将原子面A上处于4d位置的Zr原子替换为Ti原子,就形成了Zr3Ti2Si3。在原胞中,4d位置的金属原子坐标为(1/3, 2/3, 0)及其等效位置,6g位置的金属原子与硅原子坐标分别为(xM, 0, 1/4)与(xSi, 0, 1/4)及其等效位置。由于原胞中含有16个原子,这类硅化物称为16H金属硅化物。

图1 M5Si3型金属硅化物D88结构示意图
Fig.1 Schematic representation of M5Si3 silicides with D88 structure
晶格参数与弹性常数采用ABINIT[16]软件包计算。ABINIT是基于密度泛函理论及密度泛函微扰理论的平面波赝势软件包。本文的计算使用Troullier-Martins模守恒赝势[17],Ti与Zr的3d电子,Si的2s2p电子作为价电子处理。交换关联势采用Teter参数化的LDA近似。截断能量为980 eV,K点网格为4×4×6。这些参数的选择使得总能量收敛到1 meV的水平。
求出弹性常数,就可以计算其它任何弹性物理量,如体弹性模量、剪切模量、弹性模量及泊松比等。并且可以由克里斯托弗方程[18]给出沿任意方向的弹性波速。对大量不同方向的波速求平均,得到平均声速。进而根据德拜模型,由下式计算德拜温度

计算热学性质时需要先求出自由能
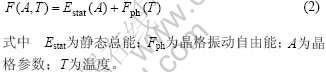
在德拜模型中,晶格振动自由能的计算进一步简化为

Fu等[11]将Estat展开为
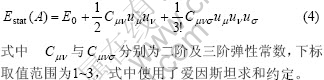
应用式(2)~(4),推出一组计算热膨胀系数的迭代公式[11]。这组迭代公式的右边分子分母均为两个与弹性常数有关的大数之差,左边为热膨胀系数这样的小量。将这组迭代公式应用于Zr5Si3及Zr3Ti2Si3热膨胀系数的计算,均无法收敛。
考虑到德拜模型已作了许多近似,将式(4)右边只取前两项,即只考虑弹性常数(二阶),忽略高阶小量,这样就直接得出德拜-格林奈森公式[19],对六方晶体结构,公式如下:

2 结果与讨论
2.1 晶格参数与重叠布居数
首先对原胞进行几何优化,计算给出0 K下晶格常数及硅原子与金属原子6g位置的坐标,列于表1。除Zr3Ti2Si3的c值的理论值与实验值相对误差约为2%外,其它晶格常数的理论值与实验值相对误差不超过1%,其中实验值为室温下的。这与通常的LDA近似下理论值与实验值的相对误差为1%左右相符。扣除温度因素,可以认为第一原理计算能够比较准确地给出平衡晶格参数。
表1 Zr5Si3与Zr3Ti2Si3晶格参数
Table 1 Lattice parameters of Zr5Si3 and Zr3Ti2Si3

在理论晶格参数下计算了原子的电子重叠布居数,列于表2,其中正、负布居数分别表明两原子间的成键与反键态[20]。布居数接近于零表明两原子间没有明显的成键,而布居数大表明形成较强的共价键。表2中,Zr―Zr (Ti―Ti) 表示两个不同的A原子面上处于4d的金属原子,Si―Zr表示同一原子面B或C的原子。
表2 Zr5Si3与Zr3Ti2Si3重叠布居
Table 2 Overlap populations for Zr5Si3and Zr3Ti2Si3
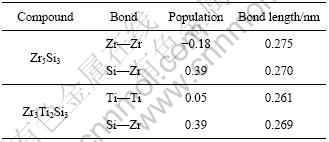
对于Zr5Si3,可以看到不同A原子面的金属原子间有负的重叠布居数,这表明反键态的存在。与之相比,同处于B或C面的原子重叠布居数为较大的正数,表明存在较强的共价键。对于Zr3Ti2Si3,原子面A上处于4d位置的Zr原子被Ti原子替换,相应的值为正数,但接近于零,而其它处于B或C面的原子重叠布居数变化很小。从成键的角度看,两种金属硅化物都是在[001]方向上的稳定性弱于(001)面上的,或者说[001]方向上的非谐效应更强,因而应该有更大的热膨胀系数。而相比之下Zr5Si3在[001]方向与(001)面上成键的差异更大一些,因而有更强的各向异性热膨胀。
2.2 弹性性质
在理论晶格参数下,计算了D88(六方)结构的弹性常数。对于六方结构共有5个独立的弹性常数,列于表3。
其中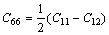 。对两种化合物都有
。对两种化合物都有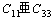 ,这表明(001)面上的原子键合作用强于[001]方向上的。
,这表明(001)面上的原子键合作用强于[001]方向上的。
表3 Zr5Si3与Zr3Ti2Si3的弹性常数
Table 3 Elastic constants of Zr5Si3 and Zr3Ti2Si3 (GPa)

从弹性常数可以计算体弹性模量、剪切模量、弹性模量及泊松比等弹性物理量,表4给出计算值。一般情况下弹性物理量的第一原理理论值与实验值的误差约为10%左右。这两种金属硅化物的弹性物理量未见有相关的实验及理论计算报道。
表4 Zr5Si3与Zr3Ti2Si3的弹性模量
Table 4 Elastic properties of Zr5Si3 and Zr3Ti2Si3

考虑到体弹性模量与剪切模量分别表征了材料抵抗断裂与塑性变形的能力,因而它们的比值K/G可作为延性或脆性的量度。比值K/G大表示延性高,比值K/G小表示脆性高。对各向同性立方晶体的计算给出K/G=1.67。作为参考,可将1.67作为表明材料延性与脆性的临界值。对Zr5Si3和Zr3Ti2Si3而言,分别有K/G=1.50和K/G=1.47。作为对比,金属Ti与Zr的体弹性模量与剪切模量比值分别为2.42和2.31。另外,较小的泊松比表明这两种金属硅化物原子间相互作用与中心力相互作用的偏离。
为考查各向异性,计算了方向体弹性模量[21]

对于Zr5Si3,Ea=Eb=290 GPa,Ec=257 GPa,Ea/Ec= 1.13。对于Zr3Ti2Si3,Ea=Eb=275 GPa,Ec=291 GPa, Ea/Ec= 0.945。以方向弹性模量为参考,Zr5Si3沿a轴方向的弹性刚度大于沿c轴方向的,而Zr3Ti2Si3则是沿a轴方向的弹性刚度略小于沿c轴方向的。
2.3 热学性质
5Si3与650 K (Zr3Ti2Si3)。根据德拜温度可以由式(7)计算定容热容。分别改变晶格参数a、c的值(1%),在新的晶格参数下计算弹性常数,并得出相应的德拜温度,由式(6)求出格林奈森常数,再由式(5)就可得出热膨胀系数与温度的关系。Zr5Si3与Zr3Ti2Si3的热膨胀理论计算值由图2给出,图中虚线为 ,实线为
,实线为 。
。
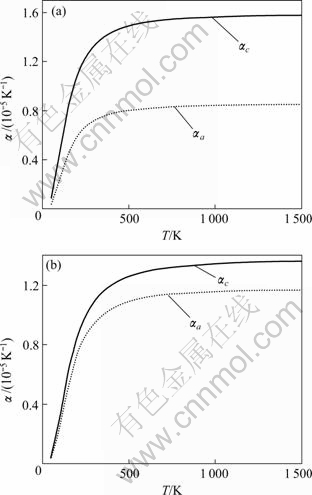
图2 热膨胀系数
Fig.2 Coefficients of thermal expansion: (a) Zr5Si3; (b) Zr3Ti2Si3
在300~1 300 K范围内,Zr5Si3的实验值为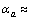
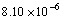 和
和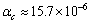 ,Zr3Ti2Si3的为
,Zr3Ti2Si3的为 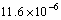 与
与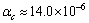 。计算结果与已有的实验数值基本符合。根据式(5),对热膨胀和弹性都是各向同性的材料,有
。计算结果与已有的实验数值基本符合。根据式(5),对热膨胀和弹性都是各向同性的材料,有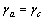 及
及 ,此时
,此时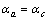 。对于Zr5Si3,
。对于Zr5Si3,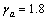 ,
,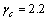 ;对于Zr3Ti2Si3,
;对于Zr3Ti2Si3,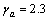 ,。格林奈森参数表征了非谐效应的大小,计算表明Zr5Si3在c轴方向的非谐效应大于在a轴方向的,而Zr3Ti2Si3在a与c方向的非谐效应相差不大。比值
,。格林奈森参数表征了非谐效应的大小,计算表明Zr5Si3在c轴方向的非谐效应大于在a轴方向的,而Zr3Ti2Si3在a与c方向的非谐效应相差不大。比值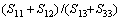 对Zr5Si3和Zr3Ti2Si3分别为1.25与1.20,表明较软的方向是[001],这也是各向异性材料里较大的热膨胀发生的方向。作为对比,金属Mg的轴比
对Zr5Si3和Zr3Ti2Si3分别为1.25与1.20,表明较软的方向是[001],这也是各向异性材料里较大的热膨胀发生的方向。作为对比,金属Mg的轴比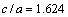 ,非常接近理想的六方密堆结构(
,非常接近理想的六方密堆结构(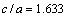 ),
),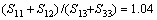 ,而两个方向的热膨胀系数比值为
,而两个方向的热膨胀系数比值为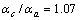 ,只有很小的各向异性。
,只有很小的各向异性。
以上对热膨胀系数的计算,晶格振动自由能不是由声子谱得出,而是将低于某一特定频率的格波作了弹性波近似,格林奈森参数也是取了平均值。另外,对静态总能的计算,只作了最低级展开。这些近似不可避免地引入一定的误差,最明显的就是热膨胀系数与定容热容cV 成比例(见式(5))。实际情况是在高温时定容摩尔热容趋于经典极限3R(R为摩尔气体常数),而热膨胀系数仍继续增大。更准确的热膨胀理论计算应该从声子谱出发,由准谐近似得出不同晶格常数下的自由能,再根据自由能极小,定出不同温度下的晶格常数,进而得出热膨胀系数与温度的关系。这种完全基于第一原理的方法计算量巨大,因而在许多时候(特别是对复杂体系)仍然需要简化的计算方案。
3 结论
1) 应用第一原理方法计算了两种金属硅化物的理论晶格参数,除Zr3Ti2Si3的c值的理论值与实验值相对误差约为2%外,其它的理论值与实验值相对误差均小于1%。
2) 计算了两种金属硅化物的弹性常数、体弹性模量、弹性模量、剪切模量及泊松比。两种金属硅化物的泊松比都较小,表明了原子间非中心力的存在。体弹性模量与剪切模量的比值K/G都小于各向同性立方晶体的相应值1.67,表明了它们有一定的脆性。C11>C33说明这两种金属硅化物在(001)面上原子间的成键作用强于[001]方向的。电荷重叠布居数的计算也得出相似的结论,位于(001)面上的原子有较强的共价键,而沿c轴方向原子间则有反键态存在。这两种键的差异,Zr5Si3比Zr3Ti2Si3表现得更大一些。
3) 对于Zr5Si3,沿a与c轴的方向体弹性模量之比为Ka/Kc=1.30,方向弹性模量之比为Ea/Ec=1.13;对Zr3Ti2Si3,相应的比值分别为Ka/Kc=1.25与Ea/Ec=0.945,这表明前者在a与c轴方向有更强的各向异性,且在a轴方向上有更大的弹性模量。从以上计算结果可以理解前者比后者有更大的各向异性热膨胀,且沿c方向热膨胀系数大于沿a方向的。
4) 应用德拜模型计算了德拜温度,分别为569 K(Zr5Si3)与650 K(Zr3Ti2Si3);对于Zr5Si3,格林奈森常数为,;对于Zr3Ti2Si3,,。这表明Zr5Si3在c轴方向的非谐效应大于在a轴方向的,而Zr3Ti2Si3在a与c方向的非谐效应相差不大。基于德拜?格林纳森模型计算了六方结构各向异性热膨胀系数,由此得出的热膨胀系数与实验基本 相符。
REFERENCES
[1] 马 勤, 阎秉钧, 康沫狂, 杨延清. 金属硅化物的应用与发展[J]. 稀有金属材料与工程, 1999, 28(1): 10?13.
MA Qin, YAN Bing-jun, KANG Mo-kuang, YANG Yan-qing. Development and applications of metal silicides[J]. Rare Metal Materials and Engineering, 1999, 28(1): 10?13.
[2] 易丹青,杜若昕,曹昱. M>sub>5Si3型硅化物的研究及相关的物理冶金学问题[J]. 金属学报, 2001, 37(11): 1121-1130.
YI Dan-qing, DU Ruo-xin, CAO Yu. Physical metallurgy of M5Si3-type silicides[J]. Acta Metallurgic Sinica, 2001, 37(11): 1121-1130.
[3] Yukinori I, Kozo I, Tamotsu N, Yutaka H, Yoshihito K. Reduction of thermal expansion anisotropy for intermetallic silicides of 16h crystal structure[J]. Intermetallics, 1996, 4(S1): S141?S145.
[4] Kagawa E, Celis P B, Ishizaki K. Design and production of the Zr3Ti2Si3intermetallic compound[J]. J Mater Res, 1991, 6(10): 2077-2083.
[5] Schneibel J H, Rawn C J, Watkins T R, Payzant E A. Thermal expansion anisotropy of ternary molybdenum silicides based on Mo5Si3[J]. Phys Rev B, 2002, 65(13): 134112?1?134112?5.
[6] Schneibel J H, Rawn C J. Thermal expansion anisotropy of ternary titanium silicides based on Ti5Si3[J]. Acta Meterialia, 2004, 52(13): 3843?3848.
[7] Schneibel J H, Rawn C J, Payzant E A, Fu C L. Controlling the thermal expansion anisotropy of Mo5Si3 and Ti5Si3 silicides[J]. Intermetallics, 2004, 12(7/9): 845?850.
[8] Fu C L, Wang X, Ye Y Y, Ho K M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation[J]. Intermetallcs, 1999, 7(2): 179?184.
[9] Narasimham S, de Gironcoli S. Ab initio calculation of the thermal properties of Cu: Performance of the LDA and GGA[J]. Phys Rev B, 2002, 65(6): 064302?1?064302?7.
[10] Wang S Q. First-principles study of the anisotropic thermal expansion of wurtzite ZnS[J]. Appl Phys Lett, 2006, 88(6): 061902?1?061902?3.
[11] Fu C L, Wang X. Thermal expansion coefficients of Mo-Si compounds by first-principles calculations[J]. Phil Mag Lett, 2000, 80(10): 683?690.
[12] Fu C L, Schneibel J H. Reducing the thermal expansion anisotropy in Mo5Si3 by Nb and V additions: theory and experiment[J]. Acta Materialia, 2003, 51(17): 5083?5092.
[13] Hayashi T, Takamoto M, Ito K, Tanaka K. The effect of Nb and W alloying additions to the thermal expansion anisotropy and elastic properties of Mo5Si3[J]. Metallurgical and Material Transactions A, 2005, 36A(3): 533?536.
[14] 孙坚, 姚强. ZrCr2 Laves 相弹性性质和堆垛层错能的第一原理计算[J]. 中国有色金属学报, 2006, 16(7): 1166?1170.
SUN Jian, YAO Qiang. First-principles study of elastic properties and stacking fault energies of ZrCr2 Laves phase[J]. The Chinese Journal of Nonferrous Metals, 2006, 16(7): 1166?1170.
[15] Mayer B, Antona H, Botta E, Methfessel M, Sticht J, Harris J, Schmidt P C. Ab initio calculation of the elastic constants and thermal expansion coeffcients of Laves phases[J]. Intermetallics, 2003, 11(12): 23?32.
[16] Gonze X, Beuken J M, Caracas R, Detraux F, Fuchs M, et al. First-principles computation of material properties: the abinit software project[J]. Computational Materials Science, 2002, 25(3): 478?492.
[17] Troullier N, Martins J. Efficient pseudopotentials for plane-wave calculations[J]. Phys Rev B, 1991, 43(3): 1993?2006.
[18] Gray D E. American institute of physics handbook[M]. Third edition. New York: McGraw-Hill Book Company, 1972.
[19] Barrera G D, Bruno J A O, Barron T H K, Allan N L. Negative thermal expansion[J]. J Phys: Condens Matter, 2005, 17(4): R217?R252.
[20] Segall M D, Shah R, Pickard C J, Payne M C. Population analysis of plane-wave electronic structure calculations of bulk materials[J]. Phys Rev B, 1996, 54(23): 16317?16320.
[21] Ravindran P, Lars F, Korzhavyi P A, Johansson B, Wills J, Eriksson O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2[J]. J Appl Phys, 1998, 84(9): 4891?4904.
基金项目:国家自然科学基金资助项目(50271085)
收稿日期:2006-12-28;修订日期:2007-06-13
通讯作者:聂耀庄,讲师,博士研究生;电话:0731-8660059;E-mail: yz_nie@163.com
(编辑 陈爱华)

