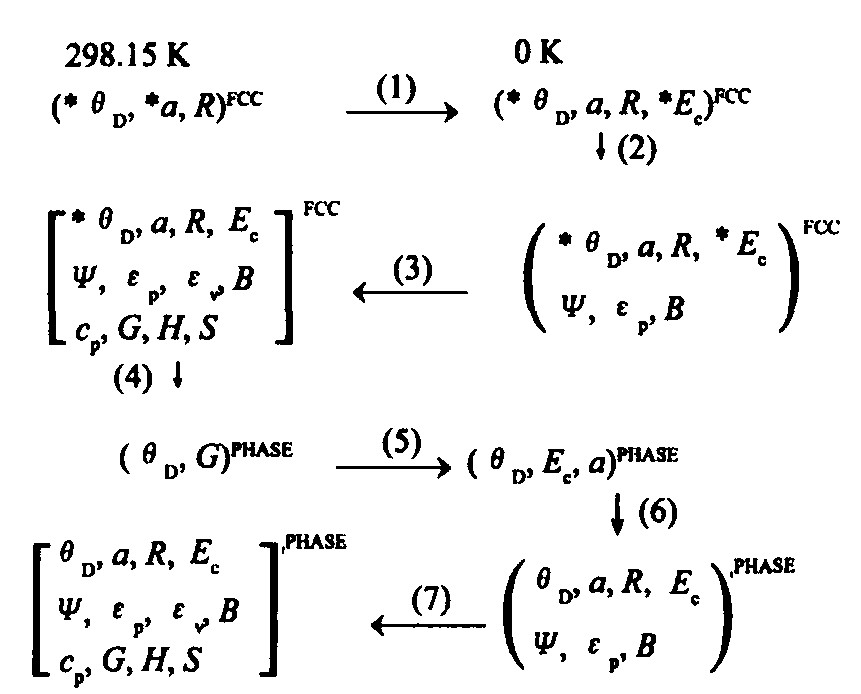稀有金属 2007,(02),206-210 DOI:10.13373/j.cnki.cjrm.2007.02.015
金属铑的原子状态、物理性质和热力学性质的研究
谢佑卿
中南大学化学化工学院,中南大学材料科学与工程学院 中南大学材料科学与工程学院,湖南长沙410083,湖南长沙410083
摘 要:
依据纯金属单原子理论 (OA) 确定了面心立方结构 (fcc) 金属Rh的原子状态为[Kr] (4dn) 4.46 (4dc) 2.54 (5sc) 1.61 (6sf) 0.39, 并对金属Rh的密排六方结构 (hcp) 和体心立方结构 (bcc) 初态特征晶体及初态液体的原子状态进行了研究, 在此基础上解释了Rh的原子状态与晶体结构的关系, 通过计算得到了fcc-Rh的势能曲线, 线热膨胀系数、晶格常数和结合能等物理性质随温度变化的曲线, 同时计算了fcc金属Rh的比热、熵、焓和Gibbs能等热力学性质随温度变化关系的曲线, 这些性质理论值与实验值符合较好, 为电催化剂及相关材料的优化设计提供了理论指导。
关键词:
金属Rh ;原子状态 ;物理性质 ;热力学性质 ;
中图分类号: TG111
收稿日期: 2006-05-17
基金: 国家自然科学基金资助项目 (50271085, 50471058); 湖南省科技计划项目 (06FJ3133);
Atomic States, Physical Properties and Thermodynamic Properties of Rhodium
Abstract:
Using the one-atom (OA) theory, the atomic state of Rh with fcc structure was determined as follows: (4dn) 4.46 (4dc) 2.54 (5sc) 1.61 (6sf) 0.39.The atomic states of Rh with hcp and bcc structure and primary liquid were also studied.According to its atomic states, the relationship between the atomic states and crystalline structure was explained qualitatively.The potential curve and the temperature dependence of linear thermal expansion coefficient, lattice constant, binding energy of fcc-Rh were calculated quantitatively.The temperature dependence of specific heat, entropy, enthalpy and Gibbs energy of fcc-Rh were calculated quantitatively.Presented data could be helpful for optimum design of Rh: based electrocatalyst and related metal material.
Keyword:
rhodium;atomic states;physical property;thermodynamic property;
Received: 2006-05-17
能源和环保是经济和社会可持续发展面临的两个主要挑战, 具有能量密度高、 无腐蚀、 工作温度低、 洁净无污染等优点的质子交换膜燃料电池 (PEMFC) 而成为研究的热点
[1 ,2 ]
。 对于PEMFC, 催化剂是决定性能、 成本的关键因素, 目前主要应用金属Pt作催化剂, 但由于金属Pt价格极其昂贵, 且资源匮乏, 使其成本居高不下, 限制了大规模的应用, 制约其商品化。 近年来研究主要围绕降低贵金属Pt担载量、 提高催化剂的稳定性和抗CO的能力及寻找价格低廉、 新型的非贵金属催化剂, 而Pt-Rh合金催化剂的形成既可降低贵金属Pt的担载量, 又可提高其稳定性及抗CO的能力
[3 ,4 ]
。 本文由纯金属系统科学, 合金物理与化学和合金统计热力学三部分组成的合金系统科学框架, 其核心内容是纯金属单原子理论 (OA)
[5 ,6 ,7 ,8 ]
和合金特征晶体理论 (CC)
[9 ,10 ,11 ,12 ]
, 依据OA理论建立的纯金属单质数据库, 对金属Rh的原子状态、 物理性质和热力学性质进行研究, 为质子交换膜燃料电池催化剂优化设计提供理论指导。
1 金属Rh的基本原子态
在OA理论中, 纯金属的原子状态以若干基本原子态φ k 组成的单原子态φ a 中准电子占有数 (QEO) 来描述:
φ a = ∑ k c k φ k ? ? ? ( 1 )
金属Rh的外层电子可分为共价电子n c 、 近自由电子n f , 非键电子n n 和磁电子n m 。 在每一基本原子态中, 电子分布服从Pauli不相容原理。
{ s c = ∑ k c k s c k ? p c = ∑ k c k p c k ? d c = ∑ k c k d c k n m = d m = ∑ k c k d m k ? n f = s f = ∑ k c k s f k ? n n = d n = ∑ k c k d n k n c = s c + p c + d c ? n v = n c + n f ? R = ∑ k c k R k ? ∑ k c k = 1 ? ? ? ( 2 )
每种基本态原子组成的赝晶体的特征性质可以通过一个与准电子数有关的多原子相互作用势能函数 (MAI势) 获得。 由晶格常数、 结合能和Debye温度的实验值, 运用单原子状态自洽法和图1所示的流程, 便可确定纯单质 (初态特征晶体) 的原子状态, 进而可以求出其有关物理性质和热力学性质, 表1为Rh的一组基本原子态及其相应的hcp , bcc 结构赝晶体的特征性质。
2 金属Rh的原子状态
在OA理论中, 以晶格常数a 和结合能E c 这两个特征性质为依据, 对14个基本态进行三态杂化组合即多种性质定态法, 确定fcc -Rh的原子状态为Ψ a (fcc -Rh) =[Kr] (4dn ) 4.46 (4dc ) 2.54 (5sc ) 1.61 (6sf ) 0.39 。
表1 金属Rh的基本原子态及其相应赝晶体的特征性质
Table 1 Basic atomic states and corresponding pseudo crystal characteristic properties of Rh metal
Number
Atomic states in outer shell
Lattice constants a /10-1 nm
Cohesive energy E c / (kJ・mol-1 )
fcc hcp * bcc fcc hcp bcc
φ 1 (4d n ) 0 (4d c ) 7 (5s c ) 2 (5s f ) 0
3.8915
2.7515
3.0969
832.03
837.30
832.49
φ 2 (4d n ) 2 (4d c ) 5 (5s c ) 2 (5s f ) 0
4.0305
2.8494
3.2307
621.70
621.91
620.86
φ 3 (4d n ) 0 (4d c ) 8 (5s c ) 0 (5s f ) 1
3.7701
2.6707
3.0923
856.51
856.55
860.90
φ 4 (4d n ) 2 (4d c ) 5 (5s c ) 1 (5s f ) 1
4.0872
2.8883
3.2535
504.72
509.23
511.45
φ 5 (4d n ) 0 (4d c ) 7 (5s c ) 1 (5s f ) 1
3.9163
2.7686
3.1377
707.18
711.07
707.05
φ 6 (4d n ) 0 (4d c ) 7 (5s c ) 0 (5s f ) 2
3.9654
2.8033
3.1776
547.60
557.85
551.45
φ 7 (4d n ) 8 (4d c ) 0 (5s c ) 0 (5s f ) 1
3.8023
2.6879
3.0469
666.93
670.90
666.38
φ 8 (4d n ) 6 (4d c ) 2 (5s c ) 1 (5s f ) 0
4.0662
3.1718
3.4976
444.34
237.19
245.18
φ 9 (4d n ) 4 (4d c ) 4 (5s c ) 1 (5s f ) 0
4.0612
2.8746
3.2367
447.26
444.51
448.73
φ 10 (4d n ) 2 (4d c ) 6 (5s c ) 1 (5s f ) 0
3.8839
2.7857
3.1120
651.24
651.49
650.53
φ 11 (4d n ) 2 (4d c ) 6 (5s c ) 0 (5s f ) 1
3.9405
2.7457
3.1359
478.90
483.13
485.18
φ 12 (4d n ) 0 (4d c ) 9 (5s c ) 0 (5s f ) 0
3.6631
2.5900
2.9155
808.39
808.43
812.11
φ 13 (4d n ) 5 (4d c ) 5 (5s c ) 0 (5s f ) 0
3.8609
2.7294
3.0720
427.40
427.56
431.37
φ 14 (4d n ) 2 (4d c ) 7 (5s c ) 0 (5s f ) 0
3.7373
2.6420
2.9916
617.89
618.10
617.39
* c /a =1.632
图1 fcc, hcp, bcc和液态Rh的原子状态和性质图 (p表示相, *表示实验值)
Fig.1 Schematic procedure for determining atomic states and properties of fcc , hcp , bcc and liquid pure Rh-metals*denotes experimental value
根据fcc -Rh的结合能E c (fcc -Rh) =554.0 kJ・mol-1 和SGTE数据库
[13 ]
给出的fcc -Rh, hcp -Rh和bcc -Rh的G ′ (T ) 值, 可近似求得hcp -Rh, bcc -Rh和L-Rh的结合能依次为: E c (hcp -Rh) =551.0 kJ・mol-1 , E c (bcc =Rh) =552.1 kJ・mol-1 , E c (L -Rh) =534.90 kJ・mol-1 。
由OA理论及图1所示的流程, 计算出hcp -Rh, bcc -Rh的晶格常数分别为: a (hcp -Rh) =0.27089 nm, a (bcc -Rh) =0.29661 nm。 确定非自然态hcp -Rh, bcc -Rh的原子状态依次为: Ψ a (hcp -Rh) =[Kr] (4dn ) 4.40 (4dc ) 2.60 (5sc ) 1.54 (5sf ) 0.46 , Ψ a (bcc -Rh) =[Kr] (4dn ) 4.42 (4dc ) 2.58 (5sc ) 1.42 (5sf ) 0.58 。
现代X射线衍射实验证实, 液态金属确实具有类似晶体的短程有序。 假定Rh在熔化后仍具有fcc 结构, 根据其在熔化前后的密度
[14 ]
(ρ s =12.42 g・cm-3 , ρ l =10.65 g・cm-3 ) 及固态fcc -Rh的晶格常数可推得L-Rh的晶格常数为a (L-Rh) =0.38597 nm。 因此L-Rh的原子状态为:Ψ a (L-Rh) =[Kr] (4dn ) 4.44 (4dc ) 2.56 (5sc ) 1.37 (5sf ) 0.63 。
3 原子状态与金属Rh晶体结构的关系
与第一原理各种方法相比, OA理论对原子状态的描述的一个重要特色是对金属的外层电子进行了功能划分, 分为共价电子、 近自由电子、 非键电子、 磁电子等, 因此可以对金属的一些性质进行定性解释。 据文献
[
15 ]
分析, 在自由原子中, d电子在未分裂的p6 壳层上运动时分成e g 和t2g 两种状态, 它们具有不同的能量和对称性。 e g 轨道沿dxyz e g 态中的d电子增加时, bcc 晶体结构趋于稳定; t2g 轨道位于px y z 2g 态中d电子增加, fcc 晶体结构趋于稳定; 在金属Rh晶体bcc -hcp -fcc 结构的转变过程中, dc 电子逐渐减少, sc 电子逐渐增加, 使电子云分布的对称性逐渐增强而方向性逐渐减弱, 形成晶体时配位数逐渐增加; 由于dc 电子和sc 电子在其结构转变过程中变化的幅度不大, 且hcp 和bcc 结构中仍含有较多的sc 电子, 这可能是hcp 和bcc -Rh难以自然存在的原因。
4 fcc-Rh物理性质的定量计算
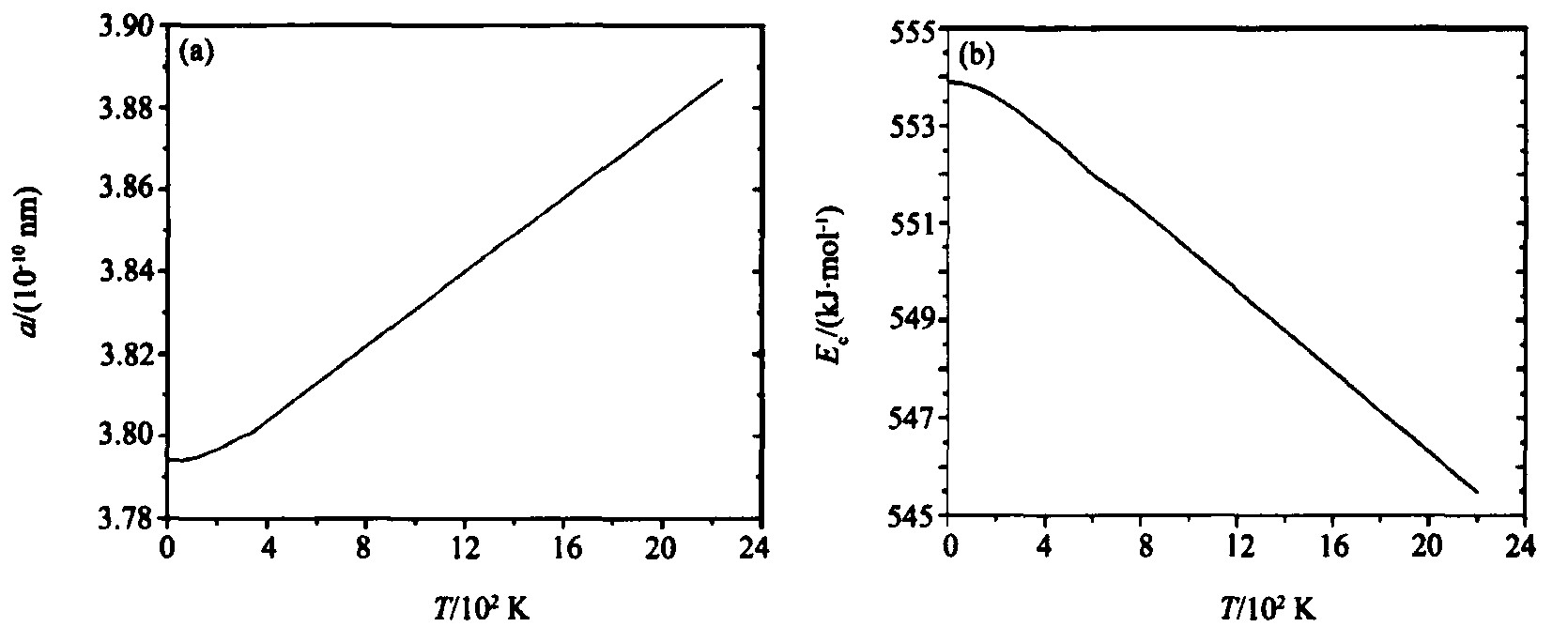
OA理论的关键是原子状态的确定, 并从电子结构层次出发对各种物理性质的定性解释和定量计算, 以期从本质上理解各种物理性质随温度的变化规律, 从而有效地加以控制; 另外这种定性解释和定量计算也可以判断得到的原子状态是否合理。 表2是fcc -Rh晶体的物理性质计算结果, 并计算了晶格常数、 结合能等物理性质随温度的变化曲线 (图2) 。
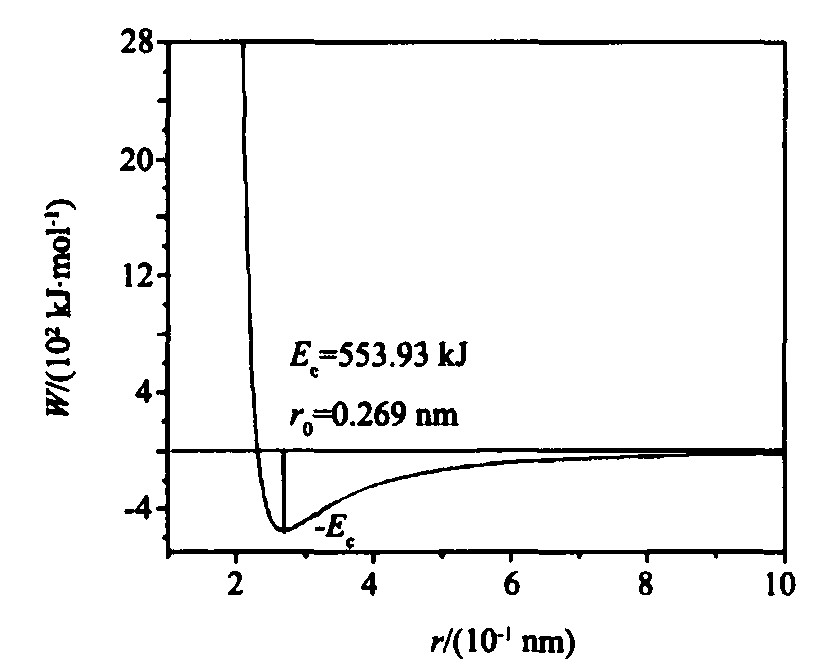
4.1 势能曲线
根据MAI势能函数
[6 ]
, 计算得到fcc -Rh的理论势能曲线如图3所示。
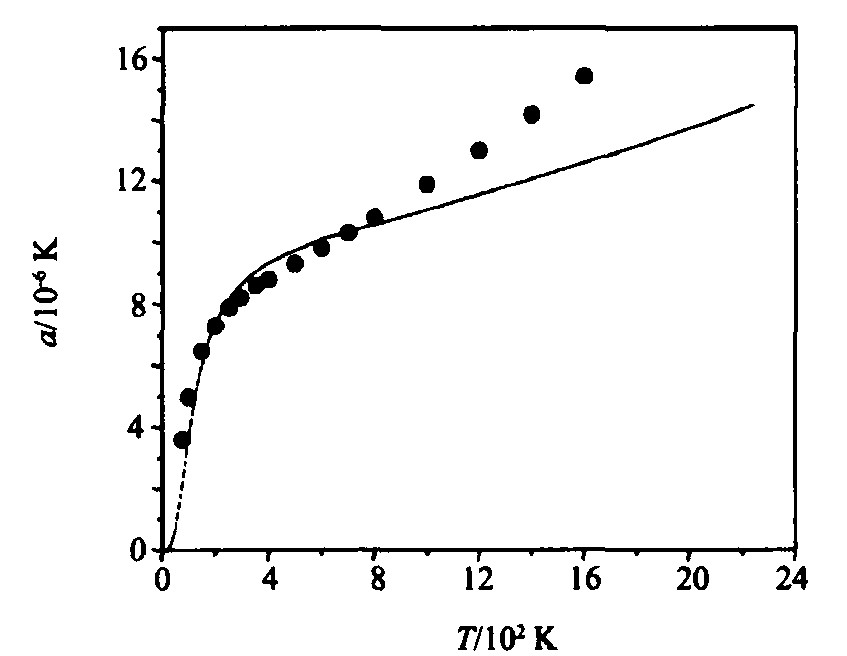
4.2 线热膨胀系数随温度的变化
根据线热膨胀系数与温度的关系式
[6 ]
, 得到fcc -Rh的线热膨胀系数随温度的变化关系曲线如图4所示, 其中Grüneisen公式中系数n =1.253, j =3.417, x =2.7046, K =3.1964, Q =869.22 kJ・mol-1 。
表2 fcc-Rh晶体的物理性质计算结果
Table 2 Calculation of physical properties of fcc Rh crystal
Atomic state
d c =2.54d n =4.46s c =1.61
Parameter
s f =0.39n c =4.15n v Τ
Bond parameters
r 1 =2.6894r 2 =3.8032r 3 =4.6483
R =1.2685n 1 =0.5572n 2 =0.0078
n 3 =0.0003
Properties
a / (10-1 nm) E c / (kJ・mol-1 ) B /GPaY /GPa
Theoretical value
3.8031
553.9
270.36
279.08
Experimental value[16]
3.8032
554.0
270.40
275.00
图2 晶格常数a (a) 和结合能 (b) 与温度变化关系的曲线
Fig.2 Lattice constants (a) and binding energy (b) as a function of temperature
图3 fcc-Rh的理论势能曲线
Fig.3 Theoretical potential curve of fcc -Rh
图4 fcc-Rh的线热膨胀系数α与温度关系的理论曲线 (・-实验值[16])
Fig.4 Linear thermal expansion coefficient as a function of temperature for fcc -Rh (・-Experimental values[16] )
图5 比热 (a) 、 熵 (b) 、 焓 (c) 和Gibbs能 (d) 与温度变化关系的曲线 (・-实验值[17])
Fig.5 Specific heat, entropy, enthalpy and Gibbs energy as a function of temperature (・-Experimental values[17] )
5 热力学性质随温度的变化
根据OA理论、 Debye-Grüneisen模型和CALPHAD (相图计算) 方法, 并按照图1所示的流程可计算fcc 金属Rh的比热c p 、 熵 (S ) 、 焓 (H ) 和Gibbs能等热力学性质随温度变化关系的曲线如图5所示, 涤图中可以看出, 其理论值与实验值符合较好, 由此也可以判断得到的原子状态是合理的。 曲线与SGTE数据库相比, 补充了其低温部分, 使曲线更加完整。 从图中可以看出, 随着温度升高, 热力学性质随着温度均发生变化, 主要是由于在升温过程中, 共价电子减少, 非键电子增加, 使得原子配位方向性减弱, 自由度增大, 电子的定域性和惰性增强, 离域性和活性减弱, 电子结构的变化是导致热力学性质发生变化的根本原因。
6 结 论
1. 确定了fcc -Rh, hcp -Rh, bcc -Rh和L-Rh的原子状态分别为Ψ a (fcc -Rh) = [Kr] (4dn ) 4.46 (4dc ) 2.54 (5sc ) 1.61 (6sf ) 0.39 ; Ψ a (hcp -Rh) =[Kr] (4dn ) 4.40 (4dc ) 2.60 (5sc ) 1.54 (5sf ) 0.46 , Ψ a (bcc -Rh) =[Kr] (4dn ) 4.42 (4dc ) 2.58 (5sc ) 1.42 (5sf ) 0.58 ; Ψ a (L-Rh) =[Kr] (4dn ) 4.44 (4dc ) 2.56 (5sc ) 1.37 (5sf ) 0.63 。
2. 依据所得到的原子状态, 定性地解释了原子状态与晶体结构的关系。
3. 根据fcc -Rh的原子状态, 计算其势能曲线, 线热膨胀系数、 晶格常数和结合能等物理性质随温度的变化曲线, 这些性质的理论值和实验值符合得较好。
4. 根据fcc-Rh的原子状态, 计算其比热、 熵、 焓和Gibbs能等热力学性质随温度变化关系, 这些性质的理论值和实验值符合得较好, 电子结构的变化是导致热力学性质发生变化的根本原因。
参考文献
[1] William H, Lizcano Valbuena, Valdecrh A, et al.Catalysts forDMFC:relation between morphology and electrochemical perfor-mance[J].Electrochimica Acta, 2003, 48 (16) :3869.
[2] 顾军, 隋升, 李光强.燃料电池中氢电极催化剂的研究[J].燃料化学学报, 1999, 27 (3) :282.
[3] Hamnett A, Kennedy B J.Bimetallic carbon supported anodes forthe direct methanol-air fuel cell[J].Electrochimica Acta, 1988, 33 (11) :1613.
[4] Koponen U, Kumpulainen H, Bergeliner M.Characterization ofPt-based catalyst materials by voltammetric techniques[J].Journalof Powder Sources, 2003, 118:325.
[5] 谢佑卿.金属材料系统科学框架[J].材料导报, 2001, 15 (4) :12.
[6] Xie Youqing.A new potential function with many-atom interactionsin solid[J].Science in China (Series A) , 1993, 36 (1) :90.
[7] Xie Youqing.One atom self-consistency method for determiningelectronic structure of crystal[J].Chinese Science Bulletin, 1992, 37 (16) :1529.
[8] Xie Youqing.Atomic energies and Gibbs energy functions of Ag-Cualloys[J].Science in China (Series E) , 1998, 41 (2) :146.
[9] Xie Youqing, Zhang Xiaodong.Phase diagram and thermodynamicproperties of Ag-Cu alloys[J].Science in China (Series E) , 1998, 41 (4) :348.
[10] Xie Youqing, Zhang Xiaodong.Atomic volumes and volume func-tions for Ag-Cu alloys[J].Science in China (Series E) , 1998, 41 (2) :157.
[11] Xie Youqing, Zhang Xiaodong.Electronic structure of Ag-Cu al-loys[J].Science in China (Series E) , 1998, 41 (3) :225.
[12] Xie Youqing, Zhang Xiaodong.Phase diagram and thermodynamicproperties of Ag-Cu alloys[J].Science in China (Series E) , 1998, 41 (4) :348.
[13] Dinsdale A T.SGTE data for pure elements[J].Calphad, 1991, 15 (4) :317.
[14] ASM International Handbook Committee.Metals handbook.10th ed, vol 2:Properties and Selection:Nonferrous Alloys and Special-Pur-pose Materials[M].OH:Materials Park, 1990.1108.
[15] 谢佑卿, 张晓东, 陈嘉砚.金属Co的原子状态和物理性质[J].中国科学 (E辑) , 1996, 28 (4) :224.
[16] American Institute of Physics.American Institute of Physics Hand-book, 3rd ed[M].New York:McGraw-Hill Book Company, 1972.4.
[17] Ralph Hultgen, Desal Pramod D, Hawkins Donald T, Molly Gleiser.Selected Values of the Thermodynamic Properties of the Elements[M].University of California, Berkeley and U.S.National Bureauof Standards, 1993.423.