
Synthesis of ��-LiV2O5/VO2 mixture by thermal lithiation of vanadium (+4, +5) oxides
LI Zhi-you(��־��), CAO Du-meng(������), ZHOU Ke-chao(�ܿƳ�)
State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China
Received 13 October 2006; accepted 28 December 2006
Abstract: ��-LiV2O5/VO2 composites were synthesized through thermal lithiation reaction of mixed valence (+4, +5) vanadium oxides by lithium bromide. The phase evolution, morphology and discharge behavior at 500 �� were investigated by thermal gravimeter/differential thermal analysis (TG/DTA), X-ray diffraction(XRD), scanning electron microscopy(SEM) and specific surface analysis(BET). The mixed vanadium oxides are obtained from the pyrolytic decomposition of ammonium metavanadate, with V6O13 as main phase. Results show that the lithiation reaction begins at about 258 ��, with ��-LiV2O5 and VO2(B) as the product. VO2(B) can transit to VO2(R) in the range of 400-500 ��, following by grain growth and crystalline development with the increase of temperature and roasting time. The ratio of ��-LiV2O5 to VO2 can be modified by the additive content of lithium bromide. A lattice shearing model about the nucleation and growth of LixV2O5 and VO2(B) inside mixed valence (+4, +5) vanadium oxides (e.g. V6O13, V3O7) is speculated, which is relative to oxygen-/vacancy-diffusion and structural evolution inspired by lithium-insertion. The open-circuit voltage of 2.6 V is observed in the single cell of Li-B/LiCl-KCl/(��-LiV2O5/VO2) at 500 ��, and the specific capacities of 146 and 167 mA?h/g (cut-off voltage 1.4 V) are measured for the positive material at 100 mA/cm2 and 200 mA/cm2, respectively.
Key words: thermal cell; ��-LiV2O5/VO2 composite material; lithiation
1 Introduction
Transition-metal oxides were investigated as the potential cathode materials of thermally activated molten salts battery. Attention was focused on the oxides based on vanadium, such as V2O5 and LiV3O8. These materials are more thermally stable and have higher open-circuit voltages than widely used disulfides (FeS2 and CoS2 mainly), but show lower usable capacities and exhibit sloping discharge curves due to frequent structure changes or insertion reactions[1-2]. The resultant product obtained by in situ thermally reacting V6O13+x with LiBr is usually entitled as LVO (lithiated vanadium oxide) that consists of ��-LiV2O5 and VO2 mainly. LVO was introduced as the cathode material of thermal cell with lithium alloys anode firstly in 1986[3]. It has more flat discharge profiles than ��-LiV2O5, VO2, their machine-made mixtures and other vanadium oxides, and is compatible with many molten electrolytes such as LiCl-KCl, LiCl-LiBr-LiF. Another material named vanadium-oxide-carbon(VOC), produced by baking the mixture of V2O5, LiCl-KCl eutectic salt and acetylene black in inert atmosphere, has the same discharge active compounds of ��-LiV2O5 and VO2 that are also from the lithiation and reduction reaction of V2O5[4-5]. Although the effects of the addition of LiBr on thermal stability and electrochemical characteristics of LVO were investigated later[6-7], any detail on the synthesis and physical performances of LVO and VOC has not been reported up to now.
The crystallographic structure of LiV2O5 and VO2 are both variable and temperature-dependent. It��s reported that VO2 has at least six polymorphs. Rutile VO2(R) (above 68 ��), monoclinic VO2(M) (below 68 ��) and triclinic VO2(T) phase are similar in structure and convertible at 52-67 ��[8]. There are other three VO2 phases designated as tetragonal VO2(A)[9], monoclinic VO2(B)[10] and layered VO2(C)[11], respectively. There are also a series of compounds LixV2O5 based on ��-V2O5 in the range of 0��x��1. The one with 0.88��x��1 is generally recorded as LiV2O5 that has at least three polymorphs denoted ��, �� and ��, respectively. ��-LiV2O5 is the high temperature phase,while �� phase appears only at low temperature and can transform spontaneously to another form �� at 110-130 ��[12-13]. But the temperature range relating to the �š��� transition is still not well defined. Several values were observed, such as 175-220 ��[12], above 300 ��[13] or 350 ��[14]. It��s not known whether any structural transformations about LiV2O5 and VO2 run in the preparation of LVO.
In this study, ��-LiV2O5/VO2 composites were in situ synthesized via thermal reaction of LiBr��H2O and vanadium oxides obtained by heating NH4VO3 in argon atmosphere. Attention was focused on the effect of synthesis condition on phase composition and resultant morphology in order to show the lithiation process. The positive discharge performances of the resultant at 500 �� were also investigated.
2 Experimental
2.1 Synthesis of ��-LiV2O5/VO2 mixed oxides
The mixed valence (+4, +5) vanadium oxides were firstly produced by calcining NH4VO3(A.R) in argon atmosphere at 450 �� for 4 h. It was then ball-milled with appropriate LiBr��H2O (A.R) in an anhydrated ethanol using a resin bottle and thpolytetrafluoroethylene balls. After dried at 120 ��, the mixtures were heated in vacuum at 320-600 �� for several hours to produce lithiated vanadium oxide.
2.2 Product characterizations
The phase compositions were evidenced by X-ray powder diffraction using a Rigaku D/max2550VB+ diffractometer with Cu K�� radiation (40 kV, 300 mA, ��= 0.154 06 nm, 10?��2�ȡ�80? or 5?��2�ȡ�85?). Thermogravimetric analysis(TGA) and differential thermal analysis(DTA) were performed in a TAS100 analyzer. Morphology of the powders was observed with a JSM-5600LV scanning electron microscope. The specific surface area of BET was measured using an ASAP2010 type instrument.
2.3 Electrochemical test of ��-LiV2O5/VO2 composite at 500 ��
The discharge process of Li-B/LiCl-KCl/(��-LiV2O5/ VO2) pellet single cell was simulated at 500 ��, and curves of voltage (vs time) were monitored by an x-y function recorder. The Li-B alloy negative with 67% Li (mass fraction, the same below) is a composite material with Li7B6 and pure Li (self-made, thickness of 0.6-0.65 mm). The electrolyte separator (mass of 0.4 g) is the powder pressed pellet of LiCl-KCl eutectic salt (with 45% LiCl, melting point 352��) and 30% porous powdered MgO binder. 30% powdered LiCl-KCl was added into ��-LiV2O5/VO2 powder (obtained at 600 �� for 4 h, the excess of LiBr��H2O was 20%), then kept at 450�� for 1 h to make LiCl-KCl be adsorbed homogeneously. After cooled and crushed, the mixture was pressed as the positive pellet (mass of 0.4 g). The diameter of electrode was 17.5 mm, and two pieces of molybdenum (thickness of 1.0 mm) were used as the collector. The preparation of electrodes and cells was performed in a glove box (relative humidity lower than 2%) to prevent the electrodes from being wetted or oxidized. The cells were loaded at several current densities to cut-off voltage of 1.4 V after being heated between two isothermal metallic tables (at 500 ��) for 1 min.
3 Results and discussion
3.1 Thermal decomposition of NH4VO3
X-ray diffraction pattern of the product from thermal decomposition of NH4VO3 at 450 �� for 4 h is shown in Fig.1. The product consists of several vanadium oxides. They are V6O13, V3O7, V2O5 and VO2(B). Similar result was found in the Ref.[3], in which V6O13, VO2 and V3O7 still co-existed even further being heated at 550 �� for 2 h. Since the thermal decomposition reaction of NH4VO3 is very complex, the decomposition process is usually described by the following two steps[1]:
2NH4VO3��V2O5+2NH3+H2O (1)
3V2O5+6NH3��V6O13+x+oxidation outcome of NH3 (2)
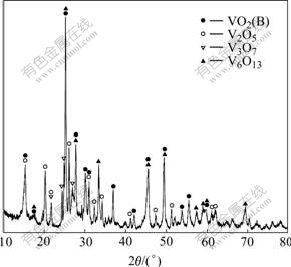
Fig.1 XRD pattern of NH4VO3 decomposition
Reactions (1) and (2) represent the decomposition of NH4VO3 and the reduction of V2O5, respectively. But the reduction reaction does not end at V6O13+x; it can react further with NH3 and the pyrolytic productions of NH3. As a result, a serial of nonstoichiometric vanadium-oxygen compounds are formed unavoidably.
The deoxidization of V2O5 to form V6O13 or VO2(B) can be described by a model of structure shear[15], in which V6O13 and VO2(B) structures are obtained from shearing the idealized V2O5 lattice by removing every third or every second (020) plane of oxygen atoms and closing the oxygen-lost polyhedra along the shear vector 1/2[110], respectively, as shown in Fig.2. V3O7 is an intermediate structure between V6O13 and V2O5, whose structure contains both single and double chains of VO6 octahedra, like V6O13, as well as some edge-shared zig-zag chains of VO5 trigonal bipyramids, as those in V2O5. By viewing of the structural intrinsic relationship, the mixture of V6O13, VO2(B) and V3O7 is produced inevitably if V2O5 is deoxidized asymmetrically. To obtain single V6O13, the molar ratio of vanadium to oxygen is needed to be controlled strictly, and the solid state reaction of stoichiometric V2O5 and vanadium is usually chosen[3,15]. Based on the structural relationship of these vanadium oxides, the product obtained at 450 �� is used directly as the vanadium oxide source to get LVO with high specific surface area.
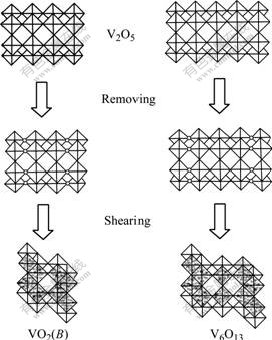
Fig.2 Sketch map of shearing ideal V2O5 crystal lattice to form VO2(B) and V6O13 lattice (Blank circles represent vacancies by removing oxygen atoms)
3.2 In situ lithiation synthesis of ��-LiV2O5/VO2 oxides
TGA and DTA curves of the mixture powder with an excess of 20% LiBr��H2O under flowing argon atmosphere are shown in Fig.3. There are two temperature ranges, in which the mass loses sharply. The first is from 80 �� to 150 ��, with the mass loss of 1.2 mg (11%), corresponding to two endothermic peaks at 118 �� and 148 �� on the DTA curve. The mass loss may be due to the evaporation of the residual balling medium, free water and bond water. The second is 150-260 ��, with 9.2% mass loss, corresponding to an exothermic peak at 258 ��. The mass loss and the heat release in this range may result from the reactions between the vanadium oxides and LiBr. It is awaited to be further confirmed whether the exothermic peak at 377 �� is related to VO2(B)��VO2(R) phase transition. This transition was observed at (440��20) ��[15], or began at 320 �� and finishes at 500 �� for nanometer VO2(B)[16]. But in our experiment the transformation isn��t completed even after being kept at 500 �� for 4 h. There is the difference of the synthesis technique and initial morphology of VO2(B) affecting the VO2(B)��VO2(R) transition. The great and broad endothermic peak at 630 �� is related to the melting of ��-LiV2O5. Because it is an incongruent melting compound, two melting-related endothermic peaks at 572 and 628 �� are once observed in a DTA study[17].
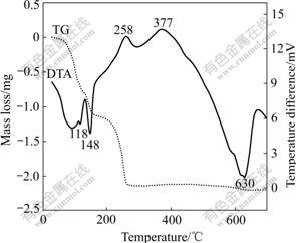
Fig.3 TG/DTA curves for vanadium oxides with LiBr��H2O (Initial mass of 11.0 mg, 10 ��/min)
The lithium-inserted structures of V2O5, V6O13 and V3O7 are changeable at high temperature. LixV6O13 is disassembled at 230-265 ��[15]. Lithium-intercalation makes V3O7 transit to lithium-rich V6O13+x even at room temperature[15], also makes V2O5 structure transform frequently. Based on their inherent stacking relationship and lithium-insertion properties, in the similar way do V6O13 and V3O7 form ��-LiV2O5/VO2(B) as V2O5. Namely, the lithiation reactions of V3O7 and V6O13 may contain two steps including the shearing breakup of the matrix resulting from the diffusion of oxygen vacancy or anion, and the nucleation and growth of new structures, as illustrated in Fig.4.
The structural micro-region of ��-V2O5 in V6O13 and V3O7 crystal is more thermodynamically advantageous to lithium-ion insertion (viz. lithiation) than that of VO2(B), which can be substantiated by the fact that V2O5 has higher electrochemical intercalation voltage than VO2(B) [18]. As lithium ions move into V2O5 micro-regions, a new structure is formed. But its frontier keeps the same structure as ��-V2O5, and shares the plane made up of double octahedral layers as the structural boundary with matrix crystal. The shear-coherence bond raises the lattice distortion near the interface, which is beneficial to breaking the double octahedral sheets. When a (020) oxygen plane in the double layers shears along 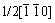 direction, a stride taking toward V6O13 or V3O7 is realized. The shear reaction is the reverse process of the transformation of V2O5 to V6O13 shown in Fig.2. The cleavage and bonding on one hand leads to the shift of the interfaces, on the other hand increases the concentration of oxygen vacancy near the interface. High oxygen vacancy concentration induces oxygen anions in V6O13 or V3O7 to configurationally transfer to the interfaces (or oxygen vacancies near the interface to the matrix phase), and in turn increases the concentration of oxygen vacancy in the matrix phase. Since a great deal of oxygen vacancy appears at VO5 micro-region ahead VO2(B) matrix, it��s helpful for the edge-shared zig-zag layers of VO5 trigonal bipyramids to undergo the shearing along 1/2[110], as shown in Fig.2. As a result, the shear drives the interfaces to move along and VO2(B) structure region spread. Vice versa, the shear that makes VO2(B) structure region spread is helpful for the extending of V2O5 structure. So the lithiation of V6O13 and V3O7 is perhaps the phenomenon that the structural decomposition is firstly induced by the diffusion of vacancy or anion, which produces the structure regions of nonstoichiometric V2O5, LixV2O5 and VO2(B), and that ��-LiV2O5 is obtained from further lithiation of LixV2O5.
direction, a stride taking toward V6O13 or V3O7 is realized. The shear reaction is the reverse process of the transformation of V2O5 to V6O13 shown in Fig.2. The cleavage and bonding on one hand leads to the shift of the interfaces, on the other hand increases the concentration of oxygen vacancy near the interface. High oxygen vacancy concentration induces oxygen anions in V6O13 or V3O7 to configurationally transfer to the interfaces (or oxygen vacancies near the interface to the matrix phase), and in turn increases the concentration of oxygen vacancy in the matrix phase. Since a great deal of oxygen vacancy appears at VO5 micro-region ahead VO2(B) matrix, it��s helpful for the edge-shared zig-zag layers of VO5 trigonal bipyramids to undergo the shearing along 1/2[110], as shown in Fig.2. As a result, the shear drives the interfaces to move along and VO2(B) structure region spread. Vice versa, the shear that makes VO2(B) structure region spread is helpful for the extending of V2O5 structure. So the lithiation of V6O13 and V3O7 is perhaps the phenomenon that the structural decomposition is firstly induced by the diffusion of vacancy or anion, which produces the structure regions of nonstoichiometric V2O5, LixV2O5 and VO2(B), and that ��-LiV2O5 is obtained from further lithiation of LixV2O5.
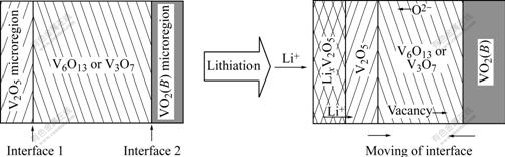
Fig.4 Schematic drawing of lithiation evolution of V3O7 and V6O13
3.3 Effect of additive content of lithiated reagent
The additive content of LiBr��H2O is calculated based on the following reaction for the preparation of lithiated vanadium oxides. The XRD patterns of composite materials with 0.8��x��1.3 in LixV2O5 are shown in Fig.5.
2LiBr��H2O+2V6O13��2LiV2O5+8VO2+Br2��+2H2O�� (3)
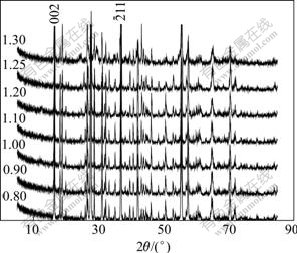
Fig.5 XRD patterns of LVO with different ratios (600 ��, 4 h) of practical mass to calculated value
The ratio of LiV2O5 to VO2 in the resultant products is reported according to the calculated value from Eqn.(3), and the optimum discharge property and the highest thermal stability can not be obtained until the additive content of LiBr is excessive[3,6-7]. To eliminate ��-Li0.13V2O5, ��-Li0.04V2O5 and V2O5 phases, 20%-30% excess of LiBr is needed[7]. However, LiV2O5/VO2 composite is gained in this experiment after 600 ��, 4 h treatment with only 80% of LiBr��H2O calculated value, in which ��-LiV2O5 content is just relatively lower and VO2 content is higher. ��-LiV2O5 content rises up and VO2 drops off with the increase of LiBr��H2O content from 0.8 to 1.3, as seen in Fig.6. Since the third intensity peak (103) of ��-LiV2O5 superposes the highest (011) peak of VO2(R), the relative contents of both are characterized by the intensity of the highest (002) peak of ��-LiV2O5 and the second high (211) peak of VO2(R), respectively. Based on the fact that ��-LiV2O5 and VO2(R) are both nonstoichiometric[15], it��s thought that ��-LiV2O5 obtained at low LiBr��H2O may be a lithium-poor phase and VO2 a oxygen-rich phase, and that the oxygen-rich VO2 can be further lithiated to form ��-LiV2O5 and oxygen-poor VO2 phase, as well as increasing lithium content in ��-LiV2O5. What is more, the lower thermal stability (decomposition temperature) of LVO obtained at low LiBr content[7] perhaps results from the transformations that lithium-poor ��-LiV2O5 separates out �¡� phase and oxide-rich VO2 separates out V2O5 phase.
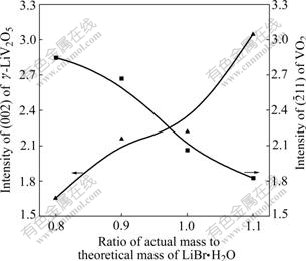
Fig.6 XRD peak intensities of ��-LiV2O5 and VO2(R) versus LiBr��H2O
3.4 Effects of experimental parameters
XRD patterns of the resultant products obtained at 320-600 �� for 4 h are shown in Fig.7. ��-LiV2O5 and VO2(B) phase are gained at 320 ��, with two unknown weak peaks at 2��=(8-9)?, which disappear above 400 ��. The resultants consist of ��-LiV2O5, VO2(B) and distinguishable VO2(R) after being heated at 400 �� and 500 ��, while ��-LiV2O5 and VO2(R) appear at 550 �� and 600 ��. It��s shown that ��-LiV2O5 and VO2(B) phases are firstly obtained from the lithiation reaction, and the metastable VO2(B) will transform gradually to the more stable rutile VO2(R) in the range of 400-550 ��.
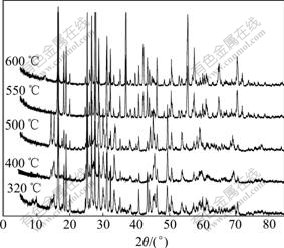
Fig.7 XRD patterns of LVO obtained at different temperatures (for 4 h)
As reported, ��-LiV2O5 can be obtained from its low-temperature counterparts, even though the required temperature varies with the lithium content, and varies greatly with measurement methods. For example, for the precursor ��-LiV2O5 prepared in an organic solution, the �š��� phase transition begins at 175-220 �� and completely finishes at 235 �� with a heating rate of 0.5 ��/min from the synchrotron X-ray powder diffraction analysis[12]. The �� phase transits quickly at 280 �� from the DSC result[19], or at 300-320 �� or 350-370 �� on the well-accepted presumable phase diagram[12, 14]. It can also be directly gained though chemical reactions, such as, hydrothermal method at 160 ��[20], organic solution processing at 120 �� using vanadium alkoxides and lithium[21], solid-state reaction of NH4V3O8 with CH3COOLi at 285.7-312 ��[22]. This observation is similar to that of the Ref.[22]. The reactions progress quickly at 258 �� and end before 280 �� (shown in Fig.3). Although it��s not quite clear whether ��-LiV2O5 nucleates and grows directly inside the vanadium oxides or comes from the transformations of its low temperature polymorphs or other low lithium phases (e.g. �� phase), it is reasonable that ��-LiV2O5 is formed though the structural transition induced by the intercalation of lithium step by step, as presumed in our model.
The lithiation reactions make the loose agglomerated particles be rock salts. The grain growth and the crystallinity characteristics are reinforced with the increase of calcination temperature and time (shown in Fig.8). The strip crystals with the radial length of less than 1 ��m and the longitudinal length of above 10 ��m come forth as bundles, and are sintered ultimately to be blocks. This indicates that the calcination, after the complete lithiation reaction, mainly enforces the growth of grain and the increasing of crystallinity except the transformation of VO2(B) to VO2(R).

Fig.8 SEM images of products obtained at different calcinated temperatures and time: (a) Vanadium oxides; (b) 500 ��, 4 h; (c) 600��, 4 h; (d) 600 ��, 2 h
The specific surface area analysis results give another evidence for the gain growth (shown in Fig.9). The specific surface area of the vanadium oxides directly from NH4VO3 is 11.26 m2/g. The values of LVO are 5.12 m2/g and 0.44 m2/g at 400 �� and 600 �� for 4 h, respectively. It also declines continuously with the prolonging of exposure time.
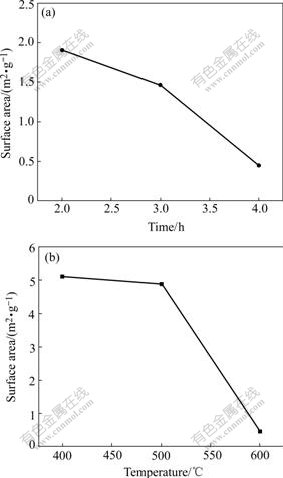
Fig.9 Specific surface area of LVO versus time (at 600 ��) (a) and temperature (for 4 h) (b)
3.5 Electrochemical test of ��-LiV2O5/VO2 composite
The discharge plots of ��-LiV2O5/VO2 material (with Li-B alloy anode at 500 ��) are shown in Fig.10. The open-circuit voltage of single thermal cell of Li-B/LiCl- KCl/(��-LiV2O5/VO2) is 2.6 V. The discharge voltage and time drop off with increasing current density. The discharge can last for 650 s till 1.4 V at the current density of 100 mA/cm2, and the specific capacity of the mixed oxides reaches 146 mA?h/g, while 167 mA?h/g at 200 mA/cm2. The discharge property is comparable to that previously reported.
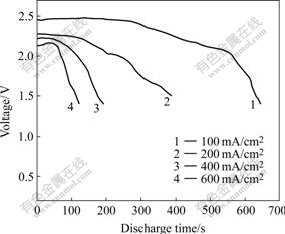
Fig.10 Electrochemical characteristics of ��-LiV2O5/VO2(R) composite
4 Conclusions
1) The product from the decomposition of ammonium metavanadate is mainly made up of V6O13, and contains minor amount of impurities such as VO2, V3O7 and V2O5. The multiphase coexistence results from the asymmetric oxygen-loss of V2O5, and does not seriously affect the phase composition of lithiated vanadium oxide.
2) The reactions between the mixed vanadium oxides and lithium bromide take place near 258 �� with ��-LiV2O5 and VO2(B) as-obtained, and VO2(B) turns to VO2(R) in the range of 400-550 ��. After the lithiation reaction, the particles grow and the crystalline morphology increases with the increased temperature and the prolonged time till the melting of ��-LiV2O5 near 630 ��. The relative content of ��-LiV2O5 and VO2 in LVO can be modified by the additive content of lithium bromide.
3) A nucleation-growth model is speculated to describe the lithiation process of mixed valence (+4, +5) vanadium oxides (e.g. V6O13, V3O7) to form LixV2O5 and VO2(B), including deoxidized shearing and oxygen- assembled shearing both inspired by lithium-insertion.
4) The open-circuit voltage of single cell of Li-B/ LiCl-KCl/(��-LiV2O5/VO2) is 2.6 V. The specific capacity of LVO reaches 146 mA?h/g and 167 mA?h/g at 100 mA/cm2 and 200 mA/cm2 till 1.4 V, respectively.
References
[1] BOLSTER M E, STANIEWICZ R J. Investigation of lithium intercalation metal oxides for thermal batteries [C]// Proceedings of the 34th International Power Sources Symposium (Cat. No.90CH2863-9). New York: IEEE, 1990: 136-140.
[2] GUIDOTTI R A, REINHARDT F W. screening study of mixed-transition-metal oxides for use as cathodes in thermal batteries [C]// Proceedings of the 37th Power Sources Conferences. Cherry Hill, NJ: Electrochemical Socieyt, Inc, 1996: 251-254.
[3] FAUL I, GOLDER A J. Electrochemical cell structures and materials therefore [P]. USA 4596752, 1986-06-24.
[4] BUCHEL J, CREPY G, DANEL V, GUIBERT A. Process for preparing a compound using a vanadium oxide derivative for the cathode of a thermal cell [P]. USA 4952467, 1990-08-28.
[5] RITCHIE A G. New cathode materials for thermal batteries [C]// The 18th International Power Sources Symposium. UK: Leatherhead, 1993: 299-312.
[6] LIU Xiao-jiang, LU Rui-sheng. Research on cathode material of lithiated vanadium oxide [J]. Chinese Journal of Power Sources, 2002, 26(1): 26-28. (in Chinese)
[7] LIU Xiao-jiang. Study on composite cathode materials for long-life thermal battery [J]. Electron Technology Reference, 2000(4): 25-31. (in Chinese)
[8] GUI Z, FAN R, CHEN X H, WU Y C. A new metastable phase of needle-like nanocrystalline VO2?H2O and phase transformation [J]. J Solid State Chemistry, 2001, 157(2): 250-254.
[9] OKA Y, YAO T, YAMAMOTO N. Powder X-ray crystal structure of VO2(A) [J]. J Solid State Chemistry, 1990, 86(1): 116-124.
[10] THEOBALD F, BERNARDS J, CABALA R. Experiment on the structure of VO2(B) [J]. J Solid State Chemistry, 1976, 17(4): 431-438.
[11] HAGRMAN D, ZUBIETA J, WARREN C J, MEYER L M, TREACY M M J, HAUSHALTER R C. A new polymorph of VO2 prepared by soft chemical methods [J]. J Solid State Chemistry, 1998, 138(1): 178-182.
[12] SATTO C, SCIAU P, DOORYHEE E, GALY J, MILLET P. The �ġ��š��� LiV2O5 ��high temperature�� phase transitions evidenced by synchrotron X-ray powder diffraction analysis [J]. J Solid State Chemistry, 1999, 146(1): 103-109.
[13] MURPY D W, CHRISTIAN P A, DISALVO F J, WASZCZAK J V. Lithium incorporation by vanadium pentoxide [J]. Inorganic Chemistry, 1979, 18(10): 2800-2803.
[14] DELMAS C, COGNAC-AURADOU H, COCCIANTELLI J M, MENETRIER M, DOUMERC J P. The LixV2O5 system: An overview of the structure modifications induced by the lithium intercalation [J]. Solid State Ionics, 1994, 69(3/4): 257-264.
[15] MURPHY D W, CHRISTIAN P A, DISALVO F J, CARIDES J N, WASZCZAK J V. Lithium incorporation by V6O13 and related vanadium (+4, +5) oxide cathode materials [J]. J Electrochem Soc, 1981, 128(10): 2053-2060.
[16] TSANG C, MANTHIRAM A. Synthesis of nanocrystalline VO2 and its electrochemical behavior in lithium batteries [J]. J Electrochem Soc, 1997, 144(2): 520-524.
[17] ERDEI S, AINGER F W. Preparation of stoichiometric �á�-LiV2O5 bronze crystals from YVO4 doped LiVO3 flux for investigation of ��"-�á� phase relation [J]. Solid State Ionics, 1994, 68(3/4): 295-304.
[18] WIESENER K, SCHNEIDER W, ILIC D, STEGER E, HALLMEIR K H, BRACKMANN E. Vanadium oxides in electrodes for rechargeable lithium cells [J]. J Power Sources, 1987, 20(1/2): 157-164.
[19] GARCIA B, MILLET M, PEREIRA-RAMOS J P, BAFFIER N, BLOCH D. Electrochemical behaviour of chemically lithiated LixV2O5 phases (0.9��x��1.6) [J]. J Power Sources, 1999, 81/82: 670-674.
[20] WANG Y W, XU H Y, WANG H, ZHANG Y C, SONG Z Q, YAN H, WAN C R. Solvothermal synthesis and characterizations of ��-LiV2O5 nanorods [J]. Solid State Ionics, 2004, 167(3/4): 419-424.
[21] OZAWA K, EGUCHI M, SAKKA Y. Low-temperature preparation of lithium vanadium oxides by solution processing [J]. J European Ceramic Society, 2004, 24(2): 405-408.
[22] DAI J X, LI S F Y, GAO Z Q, SIOW K S. Novel method for synthesis of ��-lithium vanadium oxides as cathode materials in lithium ion batteries [J]. Chem Mater, 1999, 11(11): 3086-3090.
Foundation item: Project(2003AA05510) supported by the National High-Tech Research and Development Program of China
Corresponding author: LI Zhi-you; Tel: +86-731-8830464; Fax: +86-731-8710855; E-mail: lzpm@mail.csu.edu.cn
(Edited by YANG Bing)

