���±��: 1004-0609(2005)04-0546 -06
Mg17Al12��Ca�Ͻṹ�ȶ��Եĵ�һԭ���о�
�ܵ���, �� ƽ, ׯ����, ����, ����ˮ
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ��ɳ 410082)
ժ Ҫ:���õ�һԭ������ƽ�沨����, ������Mg17Al12��Ca�Ͻ�ǰ�����̬����ӽṹ�� ��������ʾ: ��Ca�ֱ��û�Mg17Al12����Mg(��)�� Mg(��)��Mg(��)ԭ��ʱ, (Mg17-xCax)Al12�� (x=0, 1, 4, 12) �ĸ������������ߡ� �����������, ����Ca�û�Mg17Al12����Mg(��)ԭ��ʱ��Ca�Ͻ�������ǿ, �Ͻ��γɵ�(Mg5Ca12)Al12��ṹ�ȶ�����ߡ� ����̬�ܶ�(DOS)�����������: Ca�Ͻ�Mg17Al12��ṹ�ȶ������ߵ���Ҫԭ�������ںϽ�����Al(p)��Ca(s)�ļ۵���ʹ���ڵ������ijɼ����������ࡣ
�ؼ���:Mg17Al12��; ����ƽ�沨����; �����; ���ӽṹ ��ͼ�����:TG146.2
���ױ�ʶ��: A
First-principle study on structural stability of
Ca alloying Mg17Al12 phase
ZHOU Dian-wu, PENG Ping, ZHUANG Hou-long, HU Yan-jun, LIU Jin-shui
(School of Materials Science and Engineering, Hunan University, Changsha 410082, China)
Abstract: Using the first-principle pseudopotential plane-wave method, the energy and electronic structures of Ca alloying Mg17Al12 phase were investigated. The results show that the negative formation heat and the cohesive energy of (Mg17-xCax)Al12 (x=0, 1, 4, 12) phases gradually increase when the Mg atoms at ��, ��, �� positions of Mg17Al12 phase are substituted by Ca respectively, which indicates that for the alloying ability of (Mg17-xCax)Al12(x=0, 1, 4, 12) phase the replacement of Ca for Mg(��) atoms is the strongest among the above three substitutions, and the (Mg5Ca12)Al12 phase formed by this manner has the highest structural stability. After compared the densities of states (DOS) of (Mg17-xCax)Al12 phases, it is found that the increase of the structural stability of Mg17Al12 phase alloyed by Ca attributes to an increase in the bonding electron numbers at lower energy level below Feimi level, which mainly originates from the contribution of valence electron numbers of Al(p) and Ca(s) orbitals.
Key words: Mg17Al12 phase; pseudopotential plane-wave method; cohesive energy; electronic structure
þ�Ͻ���Ϲ㷺Ӧ���ں��ա� �������������ҵ�� Ӧ���������ϵ�þ�Ͻ��㲿�������ѹ����, ��Mg-Alϵ�Ͻ�ռѹ��þ�Ͻ�����90%���ϡ� �úϽ�ϵ��Ҫ�ɦ�-Mg���������-Mg17Al12���������[1], ���ڦ�-Mg17Al12���ڸ�����������, ������Ч������������Ƹ��¾���ת��, ���¸���ǿ�ȡ� ��������½�, ���������Mg-Alϵ�Ͻ��Ӧ�á� Ϊ���þ�Ͻ�ĸ����������, ͨ�����ý����������Լ��ٵ��۵㡢 ��������ֻ�Mg17Al12���ຬ��[1], ��ͨ�����ӺϽ�Ԫ����RE�� Ca�� Sr�� Si�������ɽϸ��ȶ��Ե��½����仯������ɢ��, ����CaԪ�����ڳɱ��Ͷ����ܹ�ע[2], Mg-Al-Ca�Ͻ�ϵ���ڳ�Ϊ�о��ȵ�[3-5]��
ʵ�鷢��[6]: ����Mg-Al���ȺϽ��м�������Caʱ, Ca�����϶�������Mg17Al12����, ����ʹMg17Al12����۵�ӺϽ�ǰ��449�����ߵ��˺Ͻ��468�档 ͨ��, �����仯�������ڴ��ڽ�����, �����й��ۼ������Ӽ�, ���ԭ�Ӽ�������ǿ, ���и��۵㡢 ��Ӳ�ȵ�����, ��Mg17Al12����۵�ȴ�ϵ�, Ca�Ͻ���ʹ���۵�ϴ�������, �о������û��ƽ���һ���dz�������Ĺ����� ���, ��ѧ�յ�[6]ͨ�������������(EET)�Ӽۼ��ṹ�϶��������̽��, ���ǵĹ�����Ҫ�Ǽ�����Ca��Mg17Al12���е�����Mgԭ��(��ͼ1)�û���۵��ӽṹ���Եı仯, ��ΪCa���Mg17Al12���ȶ��Ե���Ҫԭ����Ca�Ͻ������Al-Mg(��)ԭ�Ӽ�Ĺ��ۼ���ǹ��ۼ�ǿ��, ����ʹ���۵����������������ṹ�еķ�����Ӿ��ȡ� ����, ���ǽ������Եؿ����˶���Mg(��)ԭ�ӵ��û�, û�д������ĽǶȶ��䲻ͬλ��Ca�Ͻ��γ������ͺϽ���ṹ�ȶ��Եȷ����������о��� Ϊ��, �������߽�һ�����û����ܶȷ������۵ĵ�һԭ������ƽ�沨����, �Ƚ�ϵͳ���о���Mg17Al12��Ca�Ͻ�ǰ�����������ӽṹ, ����̽��Ca�Ͻ�Mg17Al12��ṹ�ȶ��Ե�Ӱ�켰��������û��ơ�
1 ����ģ���뷽��
1.1 ����ģ��
A12��Mg17Al12��ľ���ṹ��ͼ1(a)��ʾ, ������a=b=c=10.5797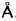 �@�� �ռ�ȺΪ
�@�� �ռ�ȺΪ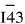 m, ��߶Գ���Ϊ
m, ��߶Գ���Ϊ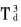 �� ������ԭ������Ϊ58, ��ԭ������Ϊ
�� ������ԭ������Ϊ58, ��ԭ������Ϊ
+2Mg(��): (0, 0, 0), (1/2, 1/2, 1/2);
+8Mg(��): (x, x, x), (-x, -x, x), x=0.32;
+24Mg(��): ( x, x, z), (-x, -x, z), (-x, x, -z), (x, -x, -z), x=0.36, z=0.04;
+24Al: (x, x, z), (-x, -x, z), (-x, x, -z), (x, -x, -z), x=0.09, z=0.28��
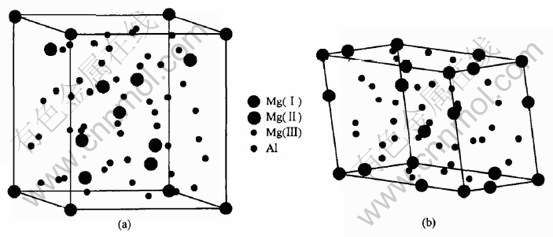
ͼ1 Mg17Al12��ľ���(a)��ԭ��(b)�ṹ
Fig.1 Modes of cell(a) and primitive cell(b) of Mg17Al12 phase
�������ԭ��ģ��, ԭ���ṹ��ͼ1 (b) ��ʾ, ԭ������Ϊ29, ����12��Alԭ�Ӻ�17��Mgԭ��(�ֱ�Ϊ1��Mg(��)�� 4��Mg(��)��12��Mg(��)ԭ��)�� ���ǵ�Caԭ����Mgԭ�Ӵ�С���ӽ�, ���Ca�Ͻ�ʱ, �ֱ���1��Ca�û�Mg17Al12ԭ���е�1��Mg(��), 4��Ca�û�4��Mg(��), 12��Ca�û�12��Mg(��), ��Ӧ�õ� (Mg16Ca)Al12�� (Mg13Ca4)Al12�� (Mg5Ca12)Al12ԭ��ģ�͡�
1.2 ���㷽��
���IJ��õļ��������Castep(Cambridge Serial Total Energy Package)���ܼ���������[7, 8], ���������������ܡ� �����ܺͽ���������3����, ���������ܲ��ù����ݶȽ���(GGA)�е�Perdew-Burke-Ernzerhof��ʽ[9, 10], ����Ϊ���ռ��еij���(ultrasoft)����[11], ������K��������ȡ 6��6��6, ���ܽضϵ�ȡ330.0eV�Ϻ����� ���������������Ǣ����(SCF)����, ��Ǣ����Ӧ��Pulay�ܶȻ�Ϸ�[12]�� ���ܼ���ʱ, ʹ���˻��������� �ڽ��и������֮ǰ, ����Broyden-Flecher-Goldfarb-Shanno (BFGS) ���������˼����Ż�, ��������ǵľ������ȶ��ṹ�� ��Ǣ����ʱ, ��ϵ������������ֵȡ5.0 ��10-6eV/ԭ��, ÿ��ԭ���ϵ�������0.01eV/�@, ����ƫ��С��5.0 ��10-4�@, Ӧ��ƫ��С��0.02GPa��
2 ���������
2.1 ƽ�⾧����
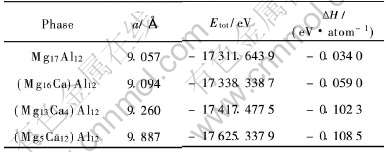
Mg17Al12�� (Mg16Ca)Al12�� (Mg13Ca4)Al12�� (Mg5Ca12)Al12ԭ����ƽ�⾧����ֵ���1��ʾ�� �����ʾ, Mg17Al12ԭ����ƽ�⾧����Ϊ 9.057�@, ��ʵ��ֵ9.145�@ (ע: ��ֵ��A12-Mg17Al12����������ʵ��ֵ10.56�@[6] ����õ�) �ܽӽ�, ���Ϊ0.962%; Ca�Ͻ�, ����ԭ����ƽ�⾧��������, ��Ca�����û�Mg17Al12���е�Mg(��)�� Mg(��)��Mg(��)ԭ��ʱ, ��Ӧ��(Mg16Ca)Al12�� (Mg13Ca4)Al12�� (Mg5Ca12)Al12ƽ�⾧������������, ����Ca�Ͻ�ʹMg17Al12��ľ����������, �˽��������[6]������Mg16.5-Ca0.5Al12�����ľ�����(Լ10.608�@)��Mg17-Al12��Ľ��һ�¡�
��1 Ca�Ͻ�ǰ��Mg17Al12���ƽ�⾧����(a)�ͺϽ�������(��H)
Table 1 Equilibrium lattice constant (a) and formation heat (��H) of Mg17Al12 phase with and without Ca addition
2.2 �Ͻ�������
�������¹�ʽ������ (Mg17-xCax)Al12ԭ��ģ��ƽ��ÿ��ԭ�ӵ�������(��H) [13, 14]:

��̬��ԭ����������ʱ, ���������ԭ����������ͬ������, Mg�� Al�� Ca���嵥ԭ�������ļ���ֵ�ֱ�Ϊ: -977.87eV�� -57.24eV�� -1003.83eV�� ����õ���Mg17Al12�� (Mg16Ca)-Al12�� (Mg13Ca4)Al12�� (Mg5Ca12)Al12�ĺϽ����������1��ʾ�� �ӱ�1�ɼ�, �û�ǰ����ĺϽ������Ⱦ�Ϊ��ֵ, ����Mg17Al12��Ca�Ͻ����γ��ȶ��ṹ[15]; ��Ca�����û���ϵ��Mg(��)�� Mg(��)��Mg(��)ԭ��ʱ, �Ͻ���������������, ��������Ca�Ͻ�Ũ������, Mg17Al12����Ca�Ͻ�������ǿ, ��Ƚ϶���, Ca�û�Mg17Al12����Mg(��)ԭ�Ӿ��б��û�Mg(��) ��Mg(��)ԭ�Ӹ�ǿ�ĺϽ��γ�������
2.3 �����
Ϊ��̽��Ca�Ͻ�Mg17Al12��ṹ�ȶ��Ե�Ӱ��, �������¹�ʽ������(Mg17-xCax)Al12ԭ���Ľ����(Ecoh)[14]:

��ͼ2�ɼ�, Ca�Ͻ�ǰ�� �ֱ��û���ϵ�е�Mg(��)�� Mg(��)��Mg(��)ԭ�Ӻ�, (Mg17-xCax)Al12ԭ����ϵ�Ľ������������ ���ھ���Ľ��ǿ��ͨ���ý��������ʾ[16], ������ܾ��ǽ�����ԭ�ӽ��Ϊ�������ͷŵ�����, Ҳ���ǰѾ���ֽ�ɵ���ԭ������Ҫ���Ĺ�, �����Խ��, �γɵľ���Խ�ȶ��� ����Mg17Al12��������ṹ�� [6], �Ͻ��γ�(Mg16Ca)Al12�� (Mg13Ca4)Al12�� (Mg5Ca12)Al12, ��ֽ������ԭ������Ҫ������Խ��, ����Ca�Ͻ�Mg17Al12��ϵ�Ľṹ�ȶ�����ǿ[17, 18], ����, Ca�û�Mg(��)ԭ��ʱ����ṹ���ȶ��� ����, �ʹӺϽ�����ѧ�۵�˵����Ϊʲô�����Mg17Al12���۵��Ca�Ͻ�ǰ��449�����ߵ��˺Ͻ��468���ʵ����[6]��ԭ��
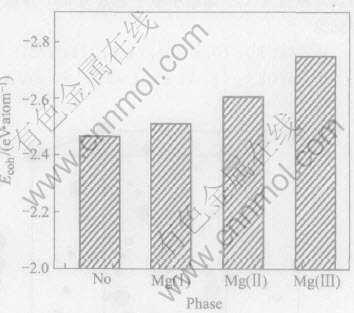
ͼ2 Ca�Ͻ�ǰ��Mg17Al12��Ľ����(Ecoh)
Fig.2 Cohesive energy (Ecoh) of Mg17Al12 phase with and without Ca addition
(No, Mg(��), Mg(��) and Mg(��) denote Mg17Al12, (Mg16Ca)Al12, (Mg13Ca4)Al12 and
(Mg5Ca12)Al12 phases, respectively)
2.4 ̬�ܶ�
Ϊ�˷���Ca�Ͻ�Mg17Al12��ṹ�ȶ������ӵĵ��ӻ���, ������Ca�Ͻ�ǰ��Mg17Al12ԭ����̬�ܶȼ���Ӧԭ�ӵķֲ�̬�ܶ�, ��������ͼ3�� �ڴ�ѡȡCa�û�Mg(��)��Mg(��)ԭ����Ϊ�о���������˷����� �û�ǰ(ͼ3(a)), ��0~-10eV��Χ��, Mg17Al12���������Ҫ�ɼ�������Mg(s)�� Al(s)��Al(p)�ļ۵��ӹ���; Ca�û�Mg(��)ԭ�Ӻ�(��ͼ3(b)), ��0~-10eV��Χ��, (Mg16Ca)Al12�����Ҫ�ɼ�����������Mg(s)�� Al(s)�� Al(p)�۵��ӵĹ���, ����-20~-25eV�ķ�Χ��, ������һ������Al(p)�۵��ӹ����³ɼ���; Ca�û�Mg(��)ԭ�Ӻ�(ͼ3(c)), ��0~-10eV��Χ��, (Mg5Ca12)Al12�����Ҫ�ɼ����ӳ�Mg(s)�� Al(s)�� Al(p) �۵��ӹ�����, ����һ��������Ca(s)�۵��ӹ���, ����-20~-25eV��Χ��, ��Ȼ��ɼ�������Ȼ����Al(p)�۵��ӵĹ���, �����ֵ��³ɼ���ĸ߶�ȴ��������
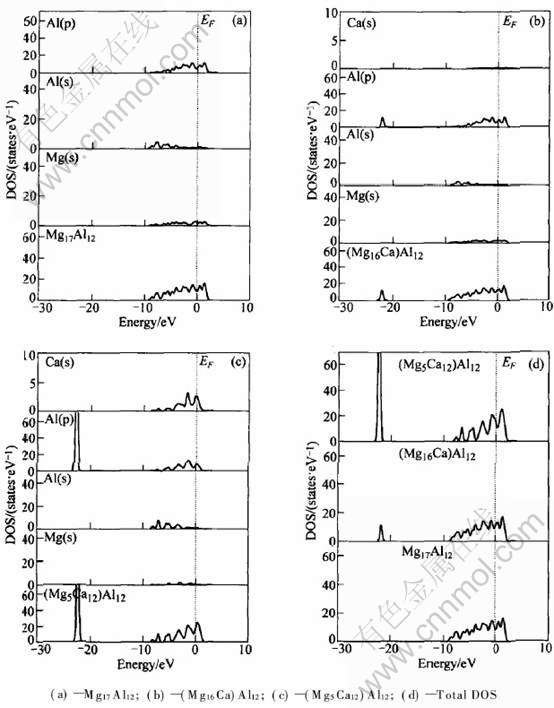
ͼ3 Ca�Ͻ�ǰ��Mg17Al12���������̬�ܶ���ֲ�̬�ܶ�
Fig.3 Total and partial DOS of Ca alloying Mg17Al12 phase
��һ���Ƚ�ͼ3(d) �ɼ�, Mg17Al12 ����Ҫ�ɼ���ֲ���0~-10eV��Χ��, Ca�û�Mg(��)ԭ�Ӻ�, (Mg16Ca)Al12��Ҫ�ɼ�������0~-10eV��Χ��, �ҳɼ���ĸ߶ȱ仯����, ����-20~-25eV��Χ��, ������һ���³ɼ���, ��ֵ�߶�Ϊ10.97������״̬/eV, ��Mg17Al12��Ca�Ͻ��ڵ��ܼ����ɼ�����������, ���Ca�û�Mg(��)ԭ�Ӻ���ϵ�ṹ��ø����ȶ�[19, 20]�� ����Ca�û�Mg(��)ԭ�Ӻ�, (Mg5Ca12)Al12����Ҫ�ɼ�����Ȼ��Ȼ��0~-10eV��Χ��, ���ɼ���ĸ߶�����, �ر�����-20~-25eV�ĵ��ܼ���Χ��, ������һ�����ߵ��³ɼ���, ��ֵ�߶ȴ�125.45������״̬/eV�� ��δ�û��Լ�Ca�û�Mg(��)ԭ�Ӻ����̬�ܶ����, ��0~-10eV��-20~-25eV�ķ�Χ��, (Mg5Ca12)Al12��ɼ��������������ࡣ ����, һ����, �ɼ����������������۵��Ӽ��������ǿ, ��һ����, ����ijɼ�����λ�ڵ��ܼ�����ʹ����ϵ�ṹ��ø����ȶ�, ���, Ca�û�Mg(��)ԭ�Ӻ���ϵ�ṹ�ͳ�Ϊ�����ȶ��Ľṹ��
3 ����
���ڵ�һԭ������ƽ�沨������Ca�Ͻ�Mg17Al12����ӽṹ�������������˼���������� �������, Mg17Al12��Ca�Ͻ���ϵ�ĸ����������ߡ� ���������, ���Ҵ�������ı仯����: ��Ca�ֱ��û�Mg17Al12����Mg(��)�� Mg(��)��Mg(��)ԭ��ʱ, (Mg17-xCax)Al12�ĸ������������ߡ� �����������, ��������Ca�Ͻ�Ũ�ȵ�����, Mg17Al12����Ca�Ͻ�������ǿ, ���ҺϽ��γɵ�(Mg16Ca)Al12�� (Mg13Ca4)Al12�� (Mg5Ca12)Al12��ṹ�ȶ��������ӡ� Ca�û�Mg17Al12����Mg(��)ԭ��ʱ, (Mg17-xCax)Al12���γ��������, ���ɵ�(Mg5Ca12)Al12��ṹ�ȶ�����ߡ� ������Ͻ�ǰ�����̬�ܶȵķ�������, Ca�Ͻ�Mg17Al12��ṹ�ȶ������ӵ���Ҫԭ����Ca�Ͻ�Mg17Al12���ڵ��ܼ����ɼ�����������, ����Դ��Ҫ��Al(p)��Ca(s)�۵��ӡ�
REFERENCES
[1] Luo A, Pekguleryuz M O. Cast magnesium alloys for elevated temperature application[J]. J Mater Sci, 1994, 29(20): 5259-5271.
[2] �κ���, Ԭ����, ������, ��. ����Mg-Zn-Si-Ca�Ͻ������֯����ѧ����[J]. �й���ɫ����ѧ��, 2002, 12(5): 956-960.
SONG Hai-ning, YUAN Guang-yin, WANG Qu-dong, et al. Microstructure and mechanical properties of heat resistant Mg-Zn-Si-Ca alloy[J]. The Chinese Journal of Nonferrous Metals, 2002, 12(5): 956-960.
[3] Min X G , Sun Y S , Xue F, et al. Analysis of valence electron structures (VES) of intermetallic compounds containing calcium in Mg-Al-based alloys[J]. Mater Chem Phys, 2002, 78(1): 88-93.
[4] Shaw C, Jones H. Structure and mechanical properties of two Mg-Al-Ca alloys consolidated from atomized power[J]. Mater Sci Tech, 1999, 15 (1): 78-83.
[5] DU Wen-wen, SUN Yang-shan, MIN Xue-gang, et al. Influence of Ca addition on valence electron structure of Mg17Al12[J]. Trans Nonferrous Met Soc China, 2003, 13(6): 1274-1280.
[6] ��ѧ��, ������, Ѧ ��, ��. Ca���Mg17Al12���۵������EET���۷���[J]. ��ѧͨ��, 2002, 47 (2): 109-112.
MIN Xue-gang, DU Wen-wen, XUE Feng, et al. The melting point of Mg17Al12 phase improved by alloyed with Ca and analysis based on the empirical electron theory (EET)[J]. Chinese Sci Bull, 2002, 47 (2): 109-112.
[7] Payne M C, Teter M P, Allan D C, et al. Iterative minization techniques for Ab initio total energy calculations: Molecular dynamics and cojugate gradients[J]. Rev Mod Phys, 1992, 64(4): 1045-1097.
[8] Lindan P J D, Segall M D, Probert M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Condens Matter, 2002, 14(11): 2717-2743.
[9] Marlo M , Milman V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals[J]. Phys Rev B, 2000, 62(4): 2899-2907.
[10] White J A , Bird D M. Implementation of gradient-corrected exchange-correlation potentials in car-par-rinello total-energy calculations[J]. Phys Rev B,1994, 50(7): 4954-4957.
[11] Vanderbilt D. Soft self-consistent pseudopotentitals in a generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41(11): 7892-7895.
[12] Hammer B, Hansen L B, Norkov J K. Improved adsorption energetics with density-functional theory using revised Perdew-Burke-Ernzerhof functionals[J]. Phys Rev B, 1999, 59(11): 7413-7421.
[13] Medvedeva M I, Gornostyrev Y N , Novikov D L, et al. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl[J]. Acta Mater, 1998, 46(10): 3433-3442.
[14] Sahu B R . Electronic structure and bonding of ultralight LiMg[J]. Mater Sci Eng B, 1997, 49(1): 74-78.
[15] Song Y, Guo Z X, Yang R, et al. First principles study of site substitution of ternary elements in NiAl[J]. Acta Mater, 2001, 49(9): 1647-1654.
[16] Zubov V I, Tretiakov N P, Teixeira Rabelo J N, et al. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a Van der Waals crystal��fullerene C60[J]. Phys Lett A, 1995, 198(5-6): 470.
[17] Ishii Y, Fujiwara T. Electronic structures and cohesion mechanism of Cd-based quasicrystals[J]. Non-cryst Solids , 2002, 312-314(12): 494-497.
[18] Fagan S B, Mota R, Baierle R J, et al. Stability investigation and thermal behavior of a hypothetical silicon nanotube[J]. J Molecular Struct, 2001, 539(1): 101-106.
[19] Fu C L, Wang X D, Ye Y Y, et al. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation[J]. Intermetallics, 1999, 7(2): 179-184.
[20] Nylen J, Garcia F J, Mosel B D, et al. Structural relationships, phase stability and bonding of compounds PdSnn (n=2, 3, 4)[J]. Solid State Sci, 2004, 6(1): 147-155.
������Ŀ: ��������ʿ�����������Ŀ(20020530012); �������Ƽ��ص�������Ŀ(104139)
�ո�����: 2004-10-27; ������: 2005-01-13
�����: �ܵ���(1971-), ��, ��ʦ, ��ʿ�о���.
ͨѶ����: �� ƽ, ��ʿ; �绰: 0731-8822663; E-mail: ppeng@hnu.cn
(�༭ Ԭ��ǰ)

