���±�ţ�1004-0609(2015)-09-2501-09
SrBi4Ti4O15����������������
ФС�죬������
(�й�ʯ�ʹ�ѧ(����) ���繤��ѧԺ���ൺ 266580)
ժ Ҫ��ͨ��ԭ�ӻ����������������SrBi4Ti4O15(SBTi)�Ŀռ����ṹ�����ݹ�������Ӿ����������(EET)����������SrBi4Ti4O15(SBTi)�ļ۵��ӽṹ��������þ����и�ԭ�ӵ���Ч�۵����������ݿռ�Ⱥ���۹���SBTi����-˳���������е�����˳��ṹ(Aama)���ķ�˳��ṹ(I4/mmm)����ͨ��ԭ����������õ��������о����ڸ�ԭ�ӵ�ԭ��λ�ơ��������Է�������ԭ��λ�ƺ�ԭ����Ч�۵�����֮��Ĺ�ϵ�����������SBTi��a�᷽����Է�����ǿ��Ϊ25.81 ��C/cm2����ʵ�������������۲ο�ֵ�ǺϽϺá�
�ؼ��ʣ��۵��ӽṹ������������ۣ�������䣻ԭ��λ�ƣ��Է�����
��ͼ����ţ�O641.1���� ���ױ�־�룺A
Ferroelectric phase transition and ferroelectricity of SrBi4Ti4O15
XIAO Xiao-hong, LI Shi-chun
(College of Mechanic and Electronic Engineering,
China University of Petroleum (East China), Qingdao 266580, China)
Abstract: The chemical bonds structures were analyzed by the atomic environment calculation method, and the valence electron structure of orthorhombic phase SrBi4Ti4O15 was calculated according to the EET theory, the numbers of the effective valence electrons of each atom in SrBi4Ti4O15 were obtained. Furthermore, according to the theory of crystal space group, the structures of orthogonal paraelectric phase (Aama) and the tetragonal paraelectric phase (I4/mmm) in the ferroelectric-paraelectric phase transition were constructed, and the atomic displacements in the phase transition were calculated by the atomic coordinate analysis. The spontaneous polarization in SrBi4Ti4O15 was studied based on the atomic displacements and atomic effective valence electrons numbers. The calculated spontaneous polarization strength in ferroelectric SrBi4Ti4O15 along the a axis is 25.81 ��C/cm2, which is in good agreement with the experimental and other theoretical results.
Key words: valence electron structure; empirical electron theory; ferroelectric phase transition; atomic displacement; spontaneous polarization
���״�ṹ������(BLSF)����AURIVILLIUS[1]��1949�귢�ֲ����ȶ�����о��������ģ��ֳ�ΪAurivillius�ࡣ�侧��ṹ������ѿ��(An-1BnO3n+1)2-��������(Bi2O2)2+��c���й���ؽ������ж��ɣ���ѧͨʽΪ(Bi2O2)2+(An-1BnO3n+1)2-[2]�����״��������Ϊһ����Ҫ����Ǧ����ѹ����ϣ������������������ܣ�������¶ȸߡ���糣��С���ϻ��ʵ͡���еƷ��������г��Ƶ���ȶ��Ժõ��ص㣬�ر������ڸ��¡���Ƶ����[3]���ڸ��´���������Ϣ�洢������й�����Ӧ��ǰ��[4-5]��
SrBi4Ti4O15(SBTi)�ǵ��͵����״�ṹ�����壬������2��(Bi2O2)2+��֮�������4������ѿ�㣬����Bλ��Tiԭ����Oԭ�ӹ���TiO6�������壬������������Ŀ�϶λ��Aλ��Sr/Biԭ��ռ�ݡ�SBTi��Ϊλ���������壬���Է������������侧��ṹ�ĶԳ���������ء��ھ����¶�����ʱSBTiΪ�����ṹ���Գ��Խϵͣ����������ԣ���Ϊ�����ࣻ���¶ȸ��ھ����¶�ʱ��SBTi�������ṹת��Ϊ�Գ��Ըߵ��ķ��ṹ���Է�������ʧ����Ϊ˳���ࡣREANEY��[6]ͨ��ʵ���ʾ��SBTi�������̲�����������ֱ�ӱ�Ϊ�ķ��࣬���Ǿ�������������-����˳��-�ķ�˳��������̡�HERVOCHES��[7]ͨ��ʵ�鷽��������ѿ����Ϊż�������״��������������ͽṹ���ܽ������о������������SBTi������-˳���������˿ռ�ȺΪA21am-Aama-I4/mmm��������У�����A21am��Aama�ṹ�IJ�ͬ������a�᷽�����Է�������λ��ģ��ʧ��
�Է�������������ı��������������������������о��ĺ������⡣KING-SMITH��[8]������ɼ۴������������Է�������Berry�����RESTA��[9]ʹ�ø÷��������˼��ѿ�������KNbO3���Է�����������һ����չ��Berry����㷽�������ǵ��о��������Է�������ԭ��λ��֮������Թ�ϵ��ͨ����ʾΪԭ��λ��������Ч��ɳ˻�֮��[10-11]�����ڸ��ѿ�ṹ��������˵�����Է�������ص����ӣ�����Ч���Ҫ��ԭ�Ӽ۴�ü��������ֵ�ɵ���������ԭ���ӻ������[12-13]����ˣ��ӵ��ӽṹ�ͼۼ����۵ĽǶ��ܹ��ܺõؽ�����������Է���������BROWN��[14]��Pauling��ѧ�����۵Ļ����ϣ������ӻ���������Ӽ�Ϊ�о�ǰ�ᣬ���ɼ�ԭ��֮��ļ���s�����d����������d0��ϵ�����������˼�������ģ�ͣ���s=exp[(d0-d)/0.37]��Brown�������ۿ�Ӧ�������Ӿ���ṹ�Ľ��ͣ�Ԥ���Լ�ԭ���������о���ͬ����Pauling��ѧ�����۵Ļ����ϣ������[15]����˹�������Ӿ����������(EET)�������۹�����ԭ�ӵĶ���ӻ�״̬��������֪�������ͨ�������� (BLD)[15-16]����õ������ڸ�ԭ�ӵ��ȶ��ӻ�״̬���۵��ӽṹ��Ϊ�����������䡢����ܡ��۵㼰��ѧ���ܵ����������ṩ�˼���Ч�ķ���[17-19]��EET���۲������������Ӿ��壬Ҳ�㷺Ӧ���ڽ������Ϻ��մɲ��ϵ������о��Ͳ������[20-23]��
�������Է������IJ���������Ҫ����������о���ṹ�Գ��ԵĽ��ͣ�������ṹ�ĶԳ���ȱ������ԭ��λ�õ�С�ı䣬��ԭ��λ�ơ����������ڲ�����ʵ��������£���SBTi�ľ���ṹ�������֣������ռ�Ⱥ���۹�����SBTi����-˳���������������ľ���ṹ��������������ṹ�仯��ԭ��λ�ơ�ͬʱ����EET����������������SBTi�ļ۵��ӽṹ���ӵ��ӽṹ�ĽǶ��о���SBTi���Է�����������ϼ۵��ӽṹ������е�ԭ��λ�Ƽ�����������SBTi���Է�����ǿ�ȡ�
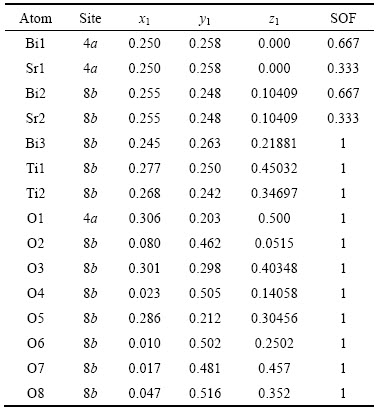
1 ����ṹ�ͼ���ֲ�
��������[7]��֪��������SBTi����Ϊ�����ṹ���ռ�ȺΪA21am(No.36)��������a=0.54507 nm��b=0.54376 nm��c= 4.09841 nm��һ�������������ġ����ӡ���Z=4������ѿ����Aλ��Bi��Sr��ԭ��ռλ����(SOF)�ֱ�Ϊ0.667��0.333������ԭ������Ϊx1��y1��z1������ṹ�������1���С�
��1 SrBi4Ti4O15�ľ���ṹ����[7]
Table 1 Crystal structure parameters of SrBi4Ti4O15[7]
������֪���������ԭ������ľ��壬ͨ��ԭ�ӻ�������[24](AEC)���Եõ�����ļ���ṹ���ݣ�����������ԭ�ӵĿռ���λ����ԭ�ӵ������λ��������һԭ��Ϊ�ο���ľ����ŴؿDz�ṹ��ԭ�ӵĿռ����˽ṹ�ȡ�������һ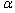 ���ĵ�ͬ����
���ĵ�ͬ���� ����ʽ(1)���[15]��
����ʽ(1)���[15]��
 (1)
(1)
ʽ�У�ISΪ�ɼ�ԭ�ӵ������λ����IMΪ�ṹ��Ԫ�ڰ����ijɼ�ԭ�ӵ���Ŀ��IKΪ���ɼ�ԭ���ڿռ�Ⱥ��ռλ����ԵIJ�����ռλ��ͬʱֵΪ1��ռλ��ͬʱֵΪ2��
��AEC�����õ���SBTi�����и�ԭ�ӵ�IS��IMֵ�������ۼ���ʵ�����d�͵�ͬ����I���ֱ����2���С�
2 ������SBTi�ļ۵��ӽṹ����
2.1 ������
������(BLD) [15-16]��EET���۵ĺ��ģ������ۻ���ΪEET������ļ��ʽ��
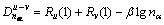 (2)
(2)
ʽ�У�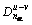 Ϊ���ࣻRu(1)��Rv(1)�ֱ�Ϊԭ��u��v�ĵ�����ࣻ�������ṹ�����в��ɺ��Եļ���n��Ϊ�����Ϸֲ��Ĺ��۵��ӶԵ���Ŀ����Ϊ������
Ϊ���ࣻRu(1)��Rv(1)�ֱ�Ϊԭ��u��v�ĵ�����ࣻ�������ṹ�����в��ɺ��Եļ���n��Ϊ�����Ϸֲ��Ĺ��۵��ӶԵ���Ŀ����Ϊ������
���ھ���ṹ��Ԫ��N����ͬ�ļ�������̼���ΪA��������һ�����䰴������С����ֱ���ΪB��C��D������N���ɼ��ʽ�õ���
 (3)
(3)
ʽ�У���עu��v��s��t�ֱ��ʾ��ͬ��ѧ���ϵ������ɼ�ԭ�ӡ�
��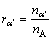 �����ݽṹ��Ԫ�ĵ�����ԭ�õ�
�����ݽṹ��Ԫ�ĵ�����ԭ�õ�
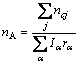 (4)
(4)
ʽ�У� Ϊ����ṹ��Ԫ��ԭ��j�Ĺ��۵�������
Ϊ����ṹ��Ԫ��ԭ��j�Ĺ��۵�������
��ʽ(3)��(4)�����ɵõ�N��n��ֵ������ֱ����N������̣����ɵõ�һ�����ۼ���ֵ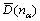 ����ѡȡ��ԭ���ӽ���ԭ�ӵ�ʵ��״̬ʱ��BLD����õ������ۼ���Ӧ��ʵ�����һ�£�һ����һ�������£�����֮��ľ���ֵС��0.005 nm�������о�������
����ѡȡ��ԭ���ӽ���ԭ�ӵ�ʵ��״̬ʱ��BLD����õ������ۼ���Ӧ��ʵ�����һ�£�һ����һ�������£�����֮��ľ���ֵС��0.005 nm�������о�������
2.2 �۵��ӽṹ����
�ڼ���ṹ���ݵĻ����ϣ�����BLD��������������SBTi�ļ۵��ӽṹ������ԭ���ӻ�״̬��[16]����Sr��7���ӻ���Bi��4���ӻ���Ti��18�ּ����ӻ���O��4���ӻ�����������㡣ԭ�ӵ��ӻ�״̬������ԭ�Ӧ��ӽ���h̬�ijɷ�Ch��t̬�ijɷ�Ct���Լ�ԭ�ӵĹ��۵�����nc�����������nl���������R(1)�Ȳ���������EET�оݣ��õ����������Ķ���ӽ���ϡ�����EET����õ���n��ֵ�Ĵ�С�ܹ���ӳ����ǿ�ȣ���ʽ�϶�Ӧ��Brown��������[14]�еļ���ֵ����ˣ��ڼ�����С��ֵ��ѡ��n��ֵ�����ֵ��Ϊ�ӽ���һ���ӽ���ϣ����ӽ�״̬����Ӧ�ļ۵��ӽṹ���2��3���С�
��2 SrBi4Ti4O15�и�ԭ���ӻ�״̬����
Table 2 Hybridization state parameters of atoms in SrBi4Ti4O15

��EET�����У���һԭ���ڽṹ��Ԫ���ṩ����Ч�۵����������Խ���Ϊ�乱���ڸ������ϵļ۵��������ܺ͡���˶���ԭ��m������Ч�۵�����qm����ͨ��ʽ(5)���н��Ƽ��㣬ԭ��m��������й��ۼ���Ҫ���е��Ӽ��㣺
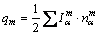 (5)
(5)
������Ҫָ�����ǣ�����ԭ��ռλ���ʲ�Ϊ1��ԭ�ӣ��ڼ���ʱӦ������ռλ���ʵ�Ӱ�졣����õ���������SBTi�����ڸ�ԭ�ӵ���Ч�۵��������4���С�
3 ��������е�ԭ��λ��
3.1 �����ķ��ṹSBTi����
SBTi���������������Ϊ����������(A21am)-����˳����(Aama)-�ķ�˳����(I4/mmm)���ο�ABRAHAMS��[25-26]��van AKEN��[27]��YMnO3�Ⱦ��������������������������SBTi�ľ���ṹ����(����1)�������������и߶Գ���ľ���ṹ��ͼ1(a)��ʾ��ϸֱ��������������������SBTi�ľ����߽��ߣ���ֱ�����ӵ���Ϊ�����ķ��ṹ������ѡ��ľ����ṹ(�����ѡ����)������ͼ(abƽ��ͼ)����ֱ�۵ر�����������������ѡ�����Ĺ�ϵ(��ͼ1(b))��
��3 SrBi4Ti4O15�ļ۵��ӽṹ
Table 3 Valence electron structure of SrBi4Ti4O15
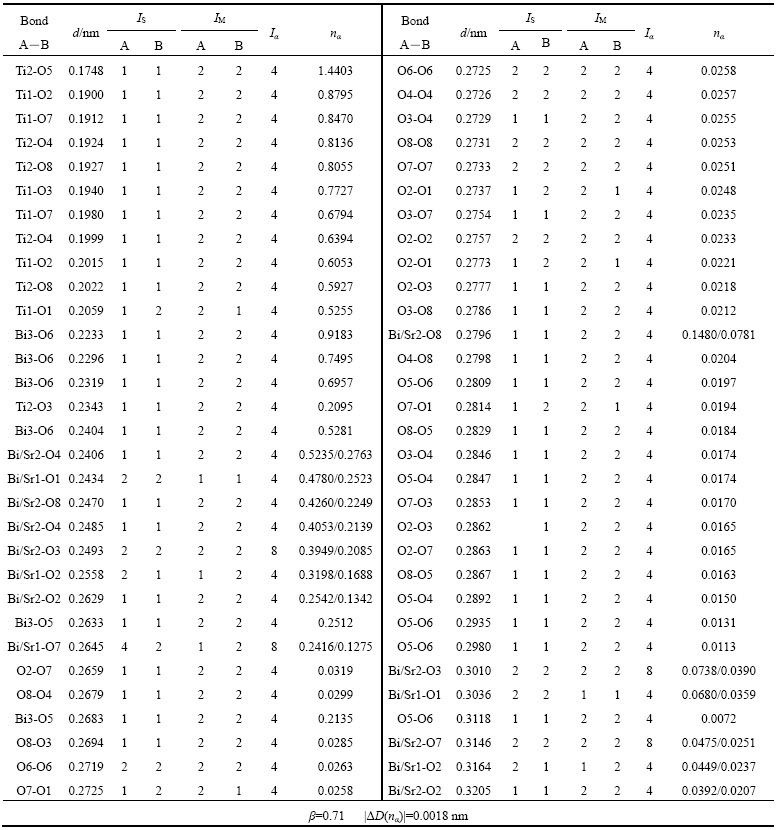
��4 SrBi4Ti4O15�и�ԭ�ӵ���Ч�۵�����
Table 4 Effective valence electrons number of SrBi4Ti4O15

Ҫ�����ķ��ṹ��SBTi���壬���Ƚ���ѡ��������ƽ�ƣ�ʹBi/Srԭ��λ������ԭ�㣬��ƽ�ƺ���ѡ�����и�ԭ�ӵ�����Ϊx1�䣬y1�䣬z1�䣬��x1��=x1-0.25��y1��=y1-0.258��z1��=z1��ͼ2��ʾΪƽ�ƺ����ѡ��������ʾ��ͼ�����ߺʹ�ʵ�������ӵĽṹ�ֱ�Ϊƽ��ǰ��AλBi/Srԭ�������γɵľ�������ṹ(c=0)��ͨ���Ա��о����֣�λ����ѡ�����������ϵ�ԭ��(��ͼ1����������Ӱ�죬��Ϊλ��mm�䣬nn�䣬oo�䣬pp�����ϵ�O2��O4��O6��O7��O8ԭ��)������������ԭ������������ѡ�����ڵ�ռλ��ͬ�������ԭ��������ƽ�ƺ��Ա��ֲ��䣬����Щԭ�ӵ�����ֵx1��=x1��y1��=y1��z1�� =z1����ѡ������a�䣬b����ֱ���Bi/Srԭ�ӵ����߷���ͼ3��ʾΪԭ������������ѡ�����ľ�������ϵ���侧�����������Խ��ߵij��ȷֱ�Ϊԭ������SBTi����ľ�����a��b�������䳤�����в��죬�����ѡ�����ľ�����a���b�䣬����Ϊ�����ṹ��
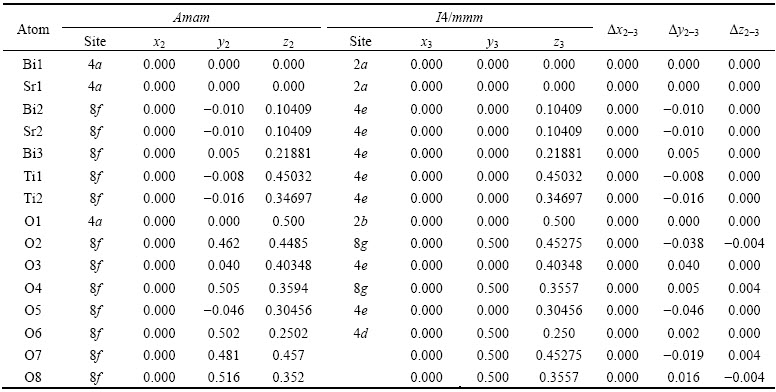
���������������˳����(Aama)��ԭ�������Ϊx2��y2��z2����ֵ�ɲ�ѯWyckoffռλ�õ���ͨ�������ǰ���ڵ�ԭ��������жԱȷ��������ɵõ���ԭ�ӵ���������������5���С���5����������Amam(No.63)��WyckoffռλΪ��4a (0,0,0)��(0��0��1/2)��8f (0��y��z)��(0��-y��z+1/2)��(0��y��-z+1/2)��(0��-y��-z)�����������������ӡ���Z=4���������������x1-2=x1��-x2����y1-2=y1��-y2����z1-2=z1��-z2���ɱ�5��ԭ�ӵ��������Կ�����������������(A21am)-����˳����(Amam)���������У�b���c�᷽����λ�ƣ�ԭ��λ����Ҫ��a�᷽����ͼ3��ԭ�����ྦྷ������ѡ�����ļ��ι�ϵ���õ�����˳����ľ�����a��=0.38542 nm��b��=0.3845 nm��c���c=4.09841 nm��������ijЩԭ�ӻ��������Сλ��ʹ��������Ϊ�ķ��ṹ�����ķ���SBTi(I4/mmm��No.139)��ԭ��������Ϊx3��y3��z3��������������������õ�����˳����(Amam)���ķ���(I4/mmm)�и�ԭ�ӵ���������(����6)����6�У��ķ����� (I4/mmm��No.139)��WyckoffռλΪ��2a (0��0��0)��2b(0��0��1/2)��4e(0��0��z)��(0��0��-z)��4d(0��1/2��1/4)��(1/2��0��1/4)��8g(0��1/2��z)��(1/2��0��z)��(0��1/2��-z)��(1/2-0��-z)�����������������ӡ���Z=2���������������x2-3=x2-x3����y2-3=y2-y3����z2-3=z2-z3��
��5 ��ѡ����������˳�����и�ԭ������
Table 5 Atomic coordinates of reselection cell and orthogonal paraelectric phase
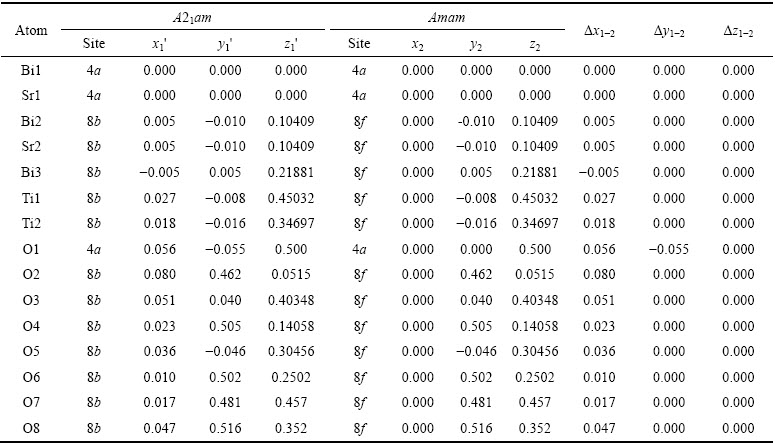
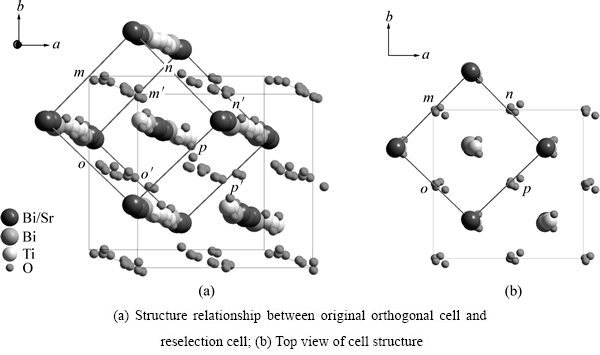
ͼ1 ԭ������������ѡ�����Ľṹͼ
Fig. 1 Structure of original orthogonal cell and reselection cell
ͨ��������������������ķ���(I4/mmm)SBTi�����У�O2 ��ͬ��������(Amam)�е�O2��O7����zֵΪ��������O2��O7��ƽ��ֵ��O4��ͬ���������е�O4��O8����zֵΪ��������O4��O8��ƽ��ֵ����ԭ�ӵ�ռλ������ֵ��������[7]�е�ʵ������һ�£����������1%��
3.2 ����ԭ��λ��
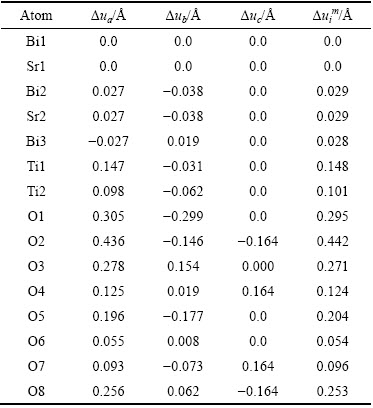
λ��������������������У��侧���ڵ�ԭ��λ�÷���ƫ�룬λ�ƵĴ�С�Ǿ����Է������������������ܵ���Ҫ����֮һ�����ݱ�5��6�и�ԭ�����ǰ������������������е�ԭ��λ�Ʀ�ua����ub�ͦ�uc [25]��������7���С�
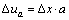 (6)
(6)
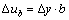 (7)
(7)
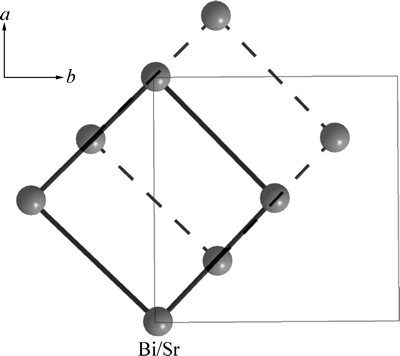
ͼ2 ƽ�ƺ����ѡ��������ʾ��ͼ
Fig. 2 Bottom surface schematic diagram of shifted reselection cell
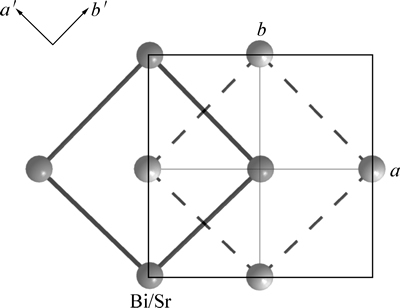
ͼ3 ԭ������������ѡ�����ľ�������ϵ
Fig. 3 Relationship of lattice constant between original orthogonal cell and reselection cell
��6 �ķ�˳����SrBi4Ti4O15��ԭ������
Table 6 Atomic coordinates of tetragonal paraelectric phase SrBi4Ti4O15
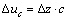 (8)
(8)
�ɱ�7�ɵã�SBTi����������������(A21am)������˳����(Amam)��Ϊ�ķ�˳����(I4/mmm)�Ĺ����У���ԭ��λ����Ҫ������abƽ�棬c�����ϵ�λ�ƺ�С�����侧����c�ɺ��Բ��ơ�ͼ4��ʾΪ�������и�ԭ����abƽ���ڵ�λ��ʸ��ͼ��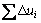 Ϊλ��ʸ���ͣ�i�������ƽ����a�����ݼ��ι�ϵ�õ���������һm��ԭ����i�����ϵ�λ�Ʒ���
Ϊλ��ʸ���ͣ�i�������ƽ����a�����ݼ��ι�ϵ�õ���������һm��ԭ����i�����ϵ�λ�Ʒ���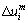 (����7)��
(����7)��
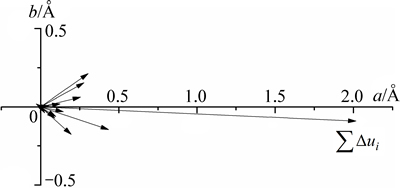
ͼ4 ԭ����abƽ���ڵ�λ��ʸ��ͼ
Fig. 4 Atomic displacement vector schematic diagram in ab plane
��7 �������е�ԭ��λ��
Table 7 Atomic displacements during phase transitions
4 ������SBTi���Է�����ǿ��
�Է���������ֱ�ӵ��������ܲ����������������������о����ص㡣IRIE��[28]ͨ��ʵ��õ�����������a(b)��ʱ��SBTi�����ı���ʣ�༫��(Pr)�ﵽ29 ��C/cm2��HERVOCHES��[7]�ڵ���ģ�͵Ļ����ϼ�����������SBTi���Է�����ǿ��(Ps)Ϊ23.2 ��C/cm2��SBTi����˳��-�����������У��ķ��ṹ�����ڵ�ԭ�ӽ��������Խ��߷�����ƫ�룬���������Խ��ߵij��Ȳ������죬�ϳ��ĶԽ��߶�Ӧ��������(A21am)�����е�a���϶̵ĶԽ��߶�Ӧ��b���Է�����������a��������ֳ������ԡ������Է�����ǿ����ԭ��λ��֮������Թ�ϵ[10-11, 22]���������߽��EET���ۣ����Լ���ǿ��(Ps)���Ʊ�ʾΪ����и�ԭ�ӵ�ԭ��λ��������Ч�۵������ij˻�֮�͡�����
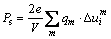 (9)
(9)
ʽ�У�Ϊ�����mԭ����i�����λ�ƣ�qmΪmԭ�ӵ���Ч�۵�������VΪ��λ���������
����4��7�е�������ݴ���ʽ(9)������õ�������SBTi������a����Է�����ǿ��Ϊ25.81 ��C/cm2��������[7]�е�����ֵ23.2 ��C/cm2�ǺϽϺá�
5 ����
1) ����EET�������ۼ�����������SBTi�ļ۵��ӽṹ����۵��ӽṹ��Sr��Bi��Ti��Oԭ���ӽֱ�Ϊ7��1��18��4���ڴ˻����ϵõ���������SBTi�и�ԭ�ӵ���Ч�۵������������ʾ�������е�Bi������TiO6����������Ti���ӵ���Ч�۵���������ԭ�Ӽ۲��ϴ�˵������ṹ������ɵ������㼰��������TiO6�ɼ�״̬�����ӽṹ�ı仯���Է������Ĺ��ϴ�
2) ���ݿռ�Ⱥ���ۣ���������SBTi�������ݵĻ����ϣ�������SBTi�������������˳����(Amam)���ķ�˳����(I4/mmm)�ľ���ṹ�����ķ���ṹ�ľ���������ԭ�ӵĿռ�ռλ���������ʵ�����ݷ��ϽϺá�
3) ͨ���������и�ԭ�ӵ�����仯�����˶�Ӧԭ�ӵ����λ�ƣ�SBTi������������ת��Ϊ�ķ���Ĺ����У���ԭ��λ��ʸ����ͺ���a�᷽���Է�����������a�᷽��ͬʱ����������֤������������(A21am)������˳����(Amam)�IJ�֮ͬ������a�᷽���ϵ�ԭ��λ�ơ�
4) �����������Է��������ۣ��������Է�����ǿ����ԭ��λ�Ƽ���Ч�۵�����֮��Ĺ�ϵʽ������õ�������SBTi��a�᷽����Է�����ǿ��Ϊ25.81 ��C/cm2����ʵ������۲ο�ֵ�ǺϽϺá�
REFERENCES
[1] AURIVILLIUS B. Mixed bismuth oxides with layer lattices. 1. The structure type of CaNb2Bi2O9[J]. Arkiv for Kemi, 1950, 1(5): 463-480.
[2] FRIT B, MERCURIO J P. The crystal chemistry and dielectric properties of the Aurivillius family of complex bismuth oxides with perovskite-like layered structures[J]. Journal of Alloys and Compounds, 1992, 188: 27-35.
[3] ������, ͯ����, ���ǿ, ������, �� ��. MBi4Ti4O15���մɵ��о���״�뷢չ����[J]. ����Ԫ�������, 2013, 32(1): 79-84.
HUANG Xin-you, TONG Xiao-feng, XU Guo-qiang, LIANG Wan-ting, TANG Hao. Research and development of MBi4Ti4O15-based ceramics[J]. Electronic Components and Materials, 2013, 32(1): 79-84.
[4] �ŷ�ǿ, ������. ���״�ṹ��������о���չ[J]. ������ѧ��, 2014, 29(5): 449-460.
ZHANG Fa-qiang, LI Yong-xiang. Recent progress on bismuth layer-structured ferroelectrics[J]. Journal of Inorganic Materials, 2014, 29(5): 449-460.
[5] ZHANG S, YU F. Piezoelectric materials for high temperature sensors[J]. Journal of the American Ceramic Society, 2011, 94(10): 3153-3170.
[6] REANEY I M, DAMJANOVIC D. Crystal structure and domain�\wall contributions to the piezoelectric properties of strontium bismuth titanate ceramics[J]. Journal of Applied Physics, 1996, 80(7): 4223-4225.
[7] HERVOCHES C H, SNEDDEN A, RIGGS R, KILCOYNE S H, MANUEL P, LIGHTFOOT P. Structural behavior of the four-layer Aurivillius-phase ferroelectrics SrBi4Ti4O15 and Bi5Ti3FeO15[J]. Journal of Solid State Chemistry, 2002, 164(2): 280-291.
[8] KING-SMITH R D, VANDERBILT D. Theory of polarization of crystalline solids[J]. Physical Review B, 1993, 47(3): 1651-1654.
[9] RESTA R, POSTERNAK M, BALDERESCHI A. Towards a quantum theory of polarization in ferroelectrics: The case of KNbO3[J]. Physical Review Letters, 1993, 70(7): 1010-1013.
[10] ��ά��. ��������ѧ[M]. ����: ��ѧ������, 1996.
ZHONG Wei-lie. Ferroelectric physics[M]. Beijing: Science Press, 1996.
[11] ABRAHAMS S C, KEVE E T. Structural basis of ferroelectricity and ferroelastcity[J]. Ferroelectrics, 1971, 2(1): 129-154.
[12] RESTA R. Macroscopic polarization in crystalline dielectrics: the geometric phase approach[J]. Reviews of Modern Physics, 1994, 66(3): 899-915.
[13] ZHONG W, VANDERBILT D, RABE K M. Phase transitions in BaTiO3 from first principles[J]. Physical Review Letters, 1994, 73(13): 1861-1864.
[14] BROWN I D, ALTERMATT D. Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database[J]. Acta Crystallographica Section B: Structural Science, 1985, 41(4): 244-247.
[15] �����. ��������Ӿ����������[J]. ��ѧͨ��, 1978, 23(4): 217-224.
YU Rui-huang. Empirical electron theory in solids and molecules[J]. Chinese Science Bulletin, 1978, 23(4): 217-224.
[16] ������. ��������Ӿ����������[M]. ����: ���ֿ�ѧ����������, 1993.
ZHANG Rui-lin. Empirical electron theory of solids and molecules[M]. Jilin: Jilin Science and Technology Press, 1993.
[17] �� ��, ��ï��, �ջԽ�, Ƚ��Ƽ. MoSi2�۵��ӽṹ����������ܼ���[J]. �й���ɫ����ѧ��, 2007, 17(2): 216-222.
PENG Ke, YI Mao-zhong, TAO Hui-jin, RAN Li-ping. Valence electronic structure analysis and cohesive energy calculation of MoSi2[J]. The Chinese Journal of Nonferrous Metals, 2007, 17(2): 216-222.
[18] ���½�, ������, �� ��. ���ѿ����������������۵��ӽṹ���ۼ���[J]. �й�ʯ�ʹ�ѧѧ��(��Ȼ��ѧ��), 2009, 33(1): 146-149.
YANG Xin-jian, LI Shi-chun, LI Hong. Theoretical calculation of valence electron structure of cubic perovskite ferroelectrics[J]. Journal of China University Petroleum (Edition of Natural Sciences), 2009, 33(1): 146-149.
[19] ��Ӣ��, �����, ����ϼ, �� ˬ, �ƴ���. Al-Mg-Si�Ͻ��ԭ�ӳɼ������ܵĹ�ϵ[J]. �й���ɫ����ѧ��, 2013, 23(5): 1226-1233.
GAO Ying-jun, CHEN Hao-tian, ZHU Tian-xia, ZHANG Shuang, HUANG Chuang-gao. Relationship between atomic bonding and property of Al-Mg-Si alloy[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(5): 1226-1233.
[20] ����Ӣ, ������. Au-Cuϵ�����仯����۵��ӽṹ���������ܼ���[J]. �й���ɫ����ѧ��, 2010, 20(4): 743-748.
JIANG Shu-ying, LI Shi-chun. Calculation of valence electron structures and cohesive energies of intermetallic compounds in Au-Cu system[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(4): 743-748.
[21] �����, ������, ��ϲ��, ������, ����Ƽ, ����ƽ, ��־ΰ. CuTi2 ��̬��Ӧ����ԭ�����ܶϼ��ĵ��������о�[J]. �й���ɫ����ѧ��, 2013, 23(5): 1282-1288.
LI Yan-ju, CHEN Yong-chong, ZHANG Xi-feng, REN Ya-kun, ZHANG Yan-ping, WANG Qiu-ping, DU Zhi-wei. Electron theory investigation on broken bonds during atomic dissolution at solid-state reaction interface of CuTi2[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(5): 1282-1288.
[22] ���½�, ������, �� ��. PbTiO3�۵��ӽṹ�������Եľ���������ۼ���[J]. ԭ�����������ѧ��, 2007, 24(B08): 140-144.
YANG Xin-jian, LI Shi-chun, LI Hong. The valence-electron structure and ferroelectricty calculation of PbTiO3 using a valence bonding electron theory[J]. Journal of Atomic and Molecular Physics, 2007, 24(B08): 140-144.
[23] ������, ֣ ��, ���ٸ�, �����. Mn��Mo2FeB2�������մ���֯����ѧ���ܵ�Ӱ��[J]. �й���ɫ����ѧ��, 2009, 19(9): 1618-1624.
PANG Xu-ming, ZHENG Yong, WANG Shao-gang, WANG Qiu-hong. Effects of Mn on structure and mechanical properties of Mo2FeB2-based cermets[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(9): 1618-1624.
[24] LI S C. AEC: A new tool for EET, TFDC and crystal formula[J]. Materials Science Forum, 2011, 689: 245-254.
[25] ABRAHAMS S C. Atomic displacements at and order of all phase transitions in multiferroic YMnO3 and BaTiO3[J]. Acta Crystallographica Section B: Structural Science, 2009, 65(4): 450-457.
[26] ABRAHAMS S C. Inorganic structures in space group P31m; coordinate analysis and systematic prediction of new ferroelectrics[J]. Acta Crystallographica Section B: Structural Science, 2010, 66(2): 173-183.
[27] van AKEN B B, MEETSMA A, PALSTRA T T M. Hexagonal YMnO3[J]. Acta Crystallographica Section C: Crystal Structure Communications, 2001, 57(3): 230-232.
[28] IRIE H, MIYAYAMA M. Dielectric and ferroelectric properties of SrBi4Ti4O15 single crystals[J]. Applied Physics Letters, 2001, 79(2): 251-253.
(�༭ �� ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(50371059)���й�ʯ�ʹ�ѧ(����)�о������¹�����Ŀ(YCX2014052)
�ո����ڣ�2015-01-29�������ڣ�2015-04-01
ͨ�����ߣ������������ڣ���ʿ���绰��0532-86983503-8320��E-mail��lishchlishch@163.com

