ОДХВұаәЕЈә1004-0609(2014)06-1408-06
ВББСОЖА©Х№РРОӘөД·ЦЧУ¶ҜБҰС§ДЈДв
БхПюІЁЈ¬РмЗмҫьЈ¬Бх ҪЈ
(ДПІэәҪҝХҙуС§ әҪҝХЦЖФм№ӨіМС§ФәЈ¬ДПІэ 330063)
ХӘ ТӘЈәІЙУГ·ЦЧУ¶ҜБҰС§·Ҫ·ЁСРҫҝВБөДБСОЖА©Х№РРОӘЎЈҪЁБўВБөД·ЦЧУ¶ҜБҰС§ДЈДвДЈРНЈ¬НЁ№эЗ¶ИлФӯЧУ·ЁҪшРРДЈДвЈ¬өГөҪВББСОЖА©Х№№эіМЦРөДФӯЧУ№мјЈНјУлДЬБҝСЭұдНјЎЈҙУБСОЖА©Х№НјЗеіюөШ№ЫІмөҪБСОЖА©Х№өДұд»ҜЗйҝцЈәБСОЖјв¶Л¶Ы»ҜЎўЧУБСОЖөДІъЙъЎўҝЧ¶ҙөДЙъіЙј°іӨҙу№эіМТФј°БСОЖәНҝЧ¶ҙөД»гјҜЎЈДЬБҝСЭұдНјЦРПкПёөШЛөГчБЛХыёцМеПөСЭұдөД№эіМЎЈМҪМЦјУФШЛЩВКәНіхКјБСОЖіӨ¶И¶ФМеПөБСОЖА©Х№РРОӘөДУ°ПмЎЈҪб№ыұнГчЈәЛжЧЕјУФШЛЩВКөДФцҙуЈ¬ФӯЧУФЛ¶ҜіМ¶ИјУҫзЈ¬МеПөНкИ«ұ»АӯҝӘөДКұјдЛх¶МЈ»іхКјБСОЖіӨ¶ИФҪіӨЈ¬ФӯЧУФЛ¶ҜөДПёҪЪұнПЦФҪІ»ГчПФЎЈ
№ШјьҙКЈәВБЈ»БСОЖА©Х№Ј»·ЦЧУ¶ҜБҰС§ДЈДвЈ»ДЬБҝСЭұдЈ»јУФШЛЩВКЈ»іхКјБСОЖіӨ¶И
ЦРНј·ЦАаәЕЈәTG146.2 ЎЎ ЎЎ ОДПЧұкЦҫВлЈәA
Molecular dynamics simulation of crack propagation behavior of aluminum
LIU Xiao-bo, XU Qing-jun, LIU Jian
(School of Aeronautical Manufacturing Engineering, Nanchang Hangkong University, Nanchang 330063, China)
Abstract: The crack propagation behavior of aluminum was studied by molecular dynamics method. The molecular dynamics simulation model of aluminum was set up, and the energy evolution map and atomic trajectory figure of aluminum crack propagation process were obtained according to the modified embedded atom method. By plotting the crack propagation graph, the change of crack propagation is observed clearly, such as the crack tip blunted, the generation of sub-crack, the formation and growth process of the void, and the collection of crack and void. The whole evolution process of the system was described in detail from energy evolution map. The effects of loading rate and initial crack length on the crack propagation behavior of the system were discussed. The results show that the degree of atomic motion is intensified and the time of system completely open is shortened with increasing the loading rate. The longer the initial crack length is, the more unobvious the details of the atomic motion are.
Key words: aluminum; crack propagation; molecular dynamics simulation; energy evolution; loading rate; initial crack length
ВБҪрКфФЪЦЖФмК№УГ№эіМЦРЈ¬І»ҝЙұЬГвөШ»біцПЦОўБСОЖЎўҝЧ¶ҙөИИұПЭЈ¬¶шХвР©ИұПЭЦрІҪСЭұдЈ¬Ҫ«өјЦВІДБПөДК§Р§ЖЖ»өЈ¬ТтҙЛЈ¬ұШРлХЖОХХвЦЦОў№ЫИұПЭөДСЭұд№жВЙЎЈ·ЦЧУ¶ҜБҰС§ДЈДвЧчОӘјЖЛг»ъДЈДвЦР·ЗіЈЦШТӘөДТ»ЦЦ·Ҫ·ЁЈ¬ФЪРн¶аБмУтЖрЧЕЦБ№ШЦШТӘөДЧчУГЈ¬УИЖдФЪГиКцОў№ЫІгҙОөДПёҪЪ·ҪГжЎЈ·ЦЧУ¶ҜБҰС§КЗЦё¶ФФӯЧУәЛәНөзЧУЛщ№№іЙөД¶аМеПөНіЈ¬ЗуҪвФЛ¶Ҝ·ҪіМЈ¬КЗТ»ЦЦДЬ№»ҪвҫцҙуБҝФӯЧУЧйіЙөДПөНі¶ҜБҰС§ОКМвөДјЖЛг·Ҫ·ЁЎЈХвЦЦ·Ҫ·ЁІ»ҪцҝЙТФЦұҪУДЈДвРн¶аОпЦКөДәк№ЫСЭұдМШРФЈ¬өГіцУлКөСйҪб№ыПа·ыәП»тПаҪьөДјЖЛгҪб№ыЈ¬¶шЗТҝЙТФМṩ΢№ЫҪб№№ЎўБЈЧУФЛ¶ҜТФј°ЛьГЗәНОпЦКәк№ЫРФЦК№ШПөөДГчИ·НјПсЈ¬ҙУ¶шОӘРВөДАнВЫәНёЕДоөД·ўХ№МṩУРБҰөДјјКхЦ§іЕЎЈ
ДҝЗ°Ј¬№ъДЪНвС§ХЯФЪБСОЖА©Х№өД·ЦЧУ¶ҜБҰС§ДЈДв·ҪГжЧцБЛҙуБҝөДСРҫҝ№ӨЧчЎЈSTALEYөИ[1]әНMACHOVAөИ[2]ІЙУГІ»Н¬өДКЖәҜКэ¶ФБСОЖГИЙъУлА©Х№ҪшРРБЛ·ЦЧУ¶ҜБҰС§ДЈДвЈ¬өГөҪБЛІ»Н¬ТтЛШ¶ФУЪБСОЖГИЙъУлА©Х№өДУ°ПмЎЈMAKI-JASKARIөИ[3]АыУГҫӯөд·ЦЧУ¶ҜБҰС§·Ҫ·ЁөДСРҫҝұнГчЈ¬НЁ№эАнПл»ҜөДДЈРНЈ¬ФЪБСОЖјв¶Л»тХЯБСОЖГИЙъҪЧ¶ОРОіЙОИ¶ЁөД»·ЧҙҪб№№Ј¬ДЬ№»ЧиЦ№БСОЖөДГИЙъЈ¬УИЖдКЗФЪФзЖЪөД¶ПБСҪЧ¶ОЎЈЦЬ№ъ»ФөИ[4]¶ФВБөҘҫ§ЦРРД№бҙ©ОўБСОЖөДУъәП№эіМҪшРРБЛ·ЦЧУ¶ҜБҰС§ДЈДвЈ¬Ҫб№ыұнГчЈ¬өұјУИИОВ¶Иі¬№эБЩҪзОВ¶И»тНвјУС№УҰБҰі¬№эБЩҪзЦөКұЈ¬ОўБСОЖҪ«НкИ«УъәПЎЈФЪБСОЖУъәП№эіМЦР°йЛжЧЕО»ҙнөДІъЙъәНФЛ¶ҜТФј°ВПҫ§әНҝХО»өДІъЙъј°ұдЗЁЎЈWEIөИ[5]ІЙУГ·ЦЧУ¶ҜБҰС§ДЈДвBCCМъІДБПОўБСОЖУъәПөДҪб№ыұнГчЈ¬ОВ¶И¶ФОўБСОЖУъәП№эіМУРПФЦшУ°ПмЎЈНхПюҫкөИ[6]К№УГИэО¬·ЦЧУ¶ҜБҰС§·Ҫ·ЁДЈДвБЛөҘҫ§ВБФӨЦЖіхКјБСОЖА©Х№№эіМЈ¬СРҫҝБЛБСОЖА©Х№»ъАнј°ОВ¶И¶ФБСОЖА©Х№№эіМөДУ°ПмЎЈWUөИ[7]ІЙУГ·ЦЧУ¶ҜБҰС§·Ҫ·ЁСРҫҝБЛОВ¶И¶ФөҘҫ§ДшІДБПБСОЖА©Х№өДУ°ПмЈ¬Ҫб№ыұнГчЈ¬БСОЖА©Х№№эіМәНУҰБҰ·ЦІјМШХчКЗГЬЗРПа№ШөДЈ¬ОВ¶ИөДұд»ҜТэЖрөҘҫ§ДшБСОЖЙъіӨЎЈөҘөВұтөИ[8]¶ФөҘҫ§НӯНдЗъөДБСОЖГИЙъәНА©Х№ҪшРРБЛ·ЦЧУ¶ҜБҰС§ДЈДвЈ¬өГөҪБЛБСОЖГИЙъәНА©Х№өДОў№Ы»ъАнЎЈ№щСЕ·јөИ[9]ФЛУГ·ЦЧУ¶ҜБҰС§ДЈДв·Ҫ·ЁСРҫҝБЛФӯЧУіЪФҘ¶ФУЪБСјвіЎөДУ°ПмЈ¬НЁ№э¶ФМеРДБў·ҪМъөДБСОЖА©Х№ҪшРРСРҫҝЈ¬ҪТКҫБЛЖдБСОЖА©Х№»ъАнЎЈCAOөИ[10]¶ФҙуҝйДЙГЧДшҫ§ ҪзЖЖБСј°БСОЖіЙәЛҪшРРБЛ·ЦЧУ¶ҜБҰС§ДЈДвЈ¬Ҫб№ыұнГчЈ¬ҫ§ҪзНСҫЫКЗДЙГЧДшҫ§өД»щұҫұдРО»ъЦЖЦ®Т»ЎЈФшПй№ъөИ[11]¶ФГҫәПҪрөДЛхҝЧәНОўБСОЖҪшРРБЛ·ЦЧУ¶ҜБҰС§ДЈДвЈ¬ҪТКҫБЛГҫәПҪрЛЬРФұдРОәНК§Р§»ъЦЖЎЈZHANGөИ[12]ФЪКТОВПВК№УГ·ЦЧУ¶ҜБҰС§ДЈДвБЛөҘҫ§UO2өД¶ПБСРРОӘЈ¬ЦӨКөБЛБСОЖјв¶ЛөДЖБұО»ъЦЖЎўО»ҙн·ўЙдәНСЗОВЧйЦҜПаөДҙ«өЭЈ¬МҪМЦБЛБСОЖјв¶ЛөДұдРО»ъЦЖЎЈХФСЮәмөИ[13]АыУГ·ЦЧУ¶ҜБҰС§·Ҫ·ЁЈ¬¶Фә¬ҝЧ¶ҙөДҪрКфНӯҪшРРАӯЙмЦБІДБП¶ПБСДЈДвСРҫҝЈ¬·ўПЦФЪҪПИхёәС№ПВЈ¬ҝЧ¶ҙј°ІДБПОӘөҜРФұдРОЈ¬і¬№эТ»¶ЁгРЦөКұіцПЦЛЬРФұдРОЈ¬ІўФЪҫЦІҝіцПЦПаұдЎЈФЪј«ЗҝөДёәС№ПВЈ¬ЛжАӯЙмУҰБҰөДФцјУЈ¬ІДБПҫӯАъөҜРФҫщФИАӯЙмЎўҫЦІҝFCCөҪHCPөДПаұдј°ИұПЭөДІъЙъЎўИұПЭ»эАЫІъЙъОўБСОЖ»тҝХ¶ҙЦұЦБІДБП¶ПБСөД№эіМЎЈЦмЦҫРЫөИ[14]ІЙУГ·ЦЧУ¶ҜБҰС§·Ҫ·Ё¶ФNi3AlәНNiAlәПҪрФЪІ»Н¬АдИҙЛЩ¶ИПВөДДэ№М№эіМҪшРРБЛСРҫҝЈ¬·ЦОцАдИҙ№эіМЦРІ»Н¬ОВ¶ИПВөДЕј·ЦІјәҜКэЎўДЬБҝәНМе»эөДұд»ҜЎЈЗсҝЛЗҝөИ[15]ІЙУГ·ЦЧУ¶ҜБҰС§ДЈДвСРҫҝБЛCu66Ti34әПҪрФЪ4ЎБ1013 K/sАдИҙЛЩ¶ИПВөДІЈБ§»ҜЧӘұдОВ¶ИәНФӯЧУөДА©ЙўРРОӘЈ¬ЦӨГчБЛУГ¶ҜБҰС§·Ҫ·ЁәНУГИИБҰС§·Ҫ·Ё»сөГөДІЈБ§ЧӘұдОВ¶ИЦ®јдөДІоТмЎЈGUOөИ[16]ІЙУГ·ЦЧУ¶ҜБҰС§·Ҫ·ЁДЈДвөҘҫ§Нӯ(100)ұнГжДЙГЧјУ№Ө№эіМЈ¬СРҫҝІДБПөДИҘіэ»ъАнәНДЙГЧјУ№Ө№эіМЦРПөНіөДОВ¶И·ЦІјУлСЭ»Ҝ№жВЙЎЈіВГчөИ[17]НЁ№э·ЦЧУ¶ҜБҰС§·Ҫ·ЁЈ¬ФЛУГEAMКЖәҜКэ·ЦұрСРҫҝБЛОЮҝЧ¶ҙәНУРҝЧ¶ҙөДДЙГЧөҘҫ§НӯёЛөДАӯЙмМШРФЎЈKARIMIөИ[18]әНTANGөИ[19]ІЙУГЗ¶ИлФӯЧУ·Ҫ·ЁЈ¬·ЦұрСРҫҝБЛөҘҫ§ДшәНөҘҫ§ГҫөДБСОЖФціӨЗйҝцЈ¬»сөГБЛБСОЖјв¶ЛөДБЩҪзёәФШәНУҰұдДЬөД·ЦІјТФј°У°ПмБСОЖФціӨөДЦШТӘТтЛШЎЈTERENTYEVөИ[20]ФЪІ»Н¬УҰұдЛЩВКЎўОВ¶ИәНБСОЖРОЧҙөДМхјюПВСРҫҝБЛBCCәНFCCЦРҪрКфБСОЖөДА©Х№әН¶Ы»ҜРРОӘЎЈҪб№ыұнГчЈ¬·ЦЧУ¶ҜБҰС§КЗСРҫҝОў№ЫБСОЖГИЙъУлА©Х№өДУРР§·Ҫ·ЁЎЈ
И»¶шЈ¬ҙу¶аКэСРҫҝХЯ¶јКЗФЪІДБПөДФӨЦЖБСОЖПВНЁ№эФӯЧУФЛ¶Ҝ№мјЈНјСРҫҝБСОЖА©Х№РРОӘөД№жВЙЎЈ¶ФУЪНЁ№эФӯЧУФЛ¶Ҝ№мјЈНјУлДЬБҝСЭұдНјПаҪбәПөД·ҪКҪАҙҪшРРБСОЖРРОӘөДСРҫҝЈ¬№ъДЪНвұЁөАәЬЙЩЎЈОӘҙЛЈ¬ұҫОДЧчХЯІЙУГEAM(Embedded-atom model)КЖәҜКэ¶ФВБҪрКфөДұдРОБСОЖА©Х№РРОӘҪшРРДЈДвЎЈІЙУГVMDИнјю¶ФХыёцБСОЖНјПсҪшРРҝЙКУ»ҜЈ¬ТФұгДЬЗеіюөШ№ЫІмБСОЖА©Х№ЗйҝцЎЈ
1 ДЈРНөДҪЁБў
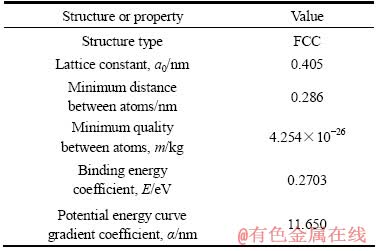
ВБОӘГжРДБў·ҪҪрКфЈ¬ЖдҪб№№ј°ОпАнМШРФИзұн1ЛщБРЎЈ
Нј1ЛщКҫОӘ·ЦЧУ¶ҜБҰС§ДЈДвөДіхКјДЈРНЎЈҙЛДЈРНПФКҫөДЗшУтҙуРЎОӘ40a0ЎБ40a0ЎБ0.25a0Ј¬a0ОӘТ»¶ЁОВ¶ИПВВБөДҫ§ёсіЈКэЎЈДЈРНМеПөЦР№ІјЖ3281ёцФӯЧУЎЈФЪXOYЖҪГжЙПЈ¬ЙПІгәНПВІг¶ФУҰөДЗшУтҙуРЎОӘ40a0ЎБ2a0Ј¬ЧуЙПІгәНЧуПВІг¶ФУҰөДЗшУтҙуРЎОӘ10a0ЎБ18a0Ј¬УТІг¶ФУҰөДЗшУтҙуРЎОӘ30a0ЎБ36a0Ј¬ФЪЧуЙПІгУлЧуПВІгФӯЧУЦ®јдЙиЦГБСОЖіӨ¶ИОӘ10a0ЎЈҪ«ЙПІгФӯЧУәНПВІгФӯЧУөДЧчУГБҰЙи¶ЁОӘ0Ј¬Ҫ«іхКјОВ¶ИЙи¶ЁОӘ1 KЈ¬ТФұЬГвФӯЧУөДИИјӨ»оР§УҰЎЈұҫДЈРНІЙУГЗ¶ИлФӯЧУ·ЁҪшРР·ЦЧУ¶ҜБҰС§ДЈДвЈ¬XәНY·ҪПтІЙУГ·ЗЦЬЖЪРФұЯҪзМхјюЈ¬Z·ҪПтІЙУГЦЬЖЪРФұЯҪзМхјюЎЈ¶ФХыёцМеПөід·ЦіЪФҘЈ¬ҙпөҪЖҪәвЧҙМ¬ЎЈёщҫЭұҫДЈРНөДҙуРЎЈ¬Ҫ«ЦРјдІгФӯЧУҙуРЎОӘ40a0ЎБ36a0өДЗшУтәНЙПІгФӯЧУЗшУтөДY·ҪПтЙПөДіхКјЛЩ¶ИЙи¶ЁОӘ30 m/sЈ¬XәНZ·ҪПтөДіхКјЛЩ¶ИЙи¶ЁОӘ0Ј¬ПВІгФӯЧУФт№М¶ЁІ»¶ҜЎЈДЈДвөДКұјдІҪіӨЙи¶ЁОӘ1ЎБ10-15 sЈ¬ХыёцМеПөІЙУГОўХэФтПөЧЫ(NVE)Ј¬ХыёціМРтФЛРР6ЎБ104ІҪЈ¬ЖЪјдГҝ1000ІҪјЗВјФӯЧУөДЧшұкО»ЦГЎў¶ҜДЬЎўКЖДЬәНЧЬДЬБҝЎЈ
ұн1 ВБөДҪб№№ј°ОпАнМШРФ
Table 1 Structure and physical properties of aluminum

Нј1 ·ЦЧУ¶ҜБҰС§ДЈДвөДіхКјДЈРН
Fig. 1 Initial model of molecular dynamics simulation
2 ·ЦЧУ¶ҜБҰС§ДЈДвөДКЖәҜКэәНЛг·Ё
2.1 КЖәҜКэДЈРН
·ЦЧУ¶ҜБҰС§өД¶ФПуКЗТ»ёцБЈЧУПөНіЈ¬ПөНіЦРФӯЧУјдөДПа»ҘЧчУГУГКЖәҜКэАҙГиКцЈ¬ТтҙЛЈ¬ХэИ·СЎФсКЖәҜКэөДАаРНј°ЖдІОКэЈ¬¶ФУЪДЈДвөДҪб№ыУЕБУҫЯУРЦШТӘЧчУГЎЈІЙУГEAMКЖәҜКэ[21]ҪшРРДЈДвЈ¬јЖЛгВБФӯЧУЦ®јдөДПа»ҘЧчУГЈ¬ПөНіөДЧЬКЖДЬұнКҫОӘ
 (1)
(1)
КҪЦРЈәFiКЗФӯЧУiөДЗ¶ИлДЬәҜКэЈ» КЗіэөЪiёцФӯЧУТФНвЛщУРФӯЧУФЪiҙҰІъЙъөДөзЧУФЖГЬ¶ИЦ®әНЈ»
КЗіэөЪiёцФӯЧУТФНвЛщУРФӯЧУФЪiҙҰІъЙъөДөзЧУФЖГЬ¶ИЦ®әНЈ» КЗөЪiёцФӯЧУУлөЪjёцФӯЧУЦ®јдөД¶ФКЖЧчУГәҜКэЈ»rijКЗөЪiёцФӯЧУУлөЪjёцФӯЧУЦ®јдөДҫаАлЎЈ
КЗөЪiёцФӯЧУУлөЪjёцФӯЧУЦ®јдөД¶ФКЖЧчУГәҜКэЈ»rijКЗөЪiёцФӯЧУУлөЪjёцФӯЧУЦ®јдөДҫаАлЎЈ
2.2 ДЈДвІЙУГөДЛЩ¶ИЛг·Ё
ІЙУГVelocity-VerletЛг·ЁЈ¬ёГЛг·ЁёшіцЛЩ¶ИЎўјУЛЩ¶ИәНФӯЧУөДО»ЦГЈ¬ІўЗТІ»ҪөөНҫ«¶ИЈ¬»№ёшіцБЛПФКҪЛЩ¶ИПоЈ¬јЖЛгБҝККЦРЈ¬УҰУГҪП№г·әЎЈјҙ
 (2)
(2)
 (3)
(3)
УЙКҪ(2)әН(3)ҝЙНЖөјіцКҪ(4)әН(5)Јә
 (4)
(4)
 (5)
(5)
КҪЦРЈәrКЗФӯЧУөДО»ЦГЧшұкЈ»vКЗФӯЧУөДЛЩ¶ИЈ»aКЗФӯЧУөДјУЛЩ¶ИЈ»tКЗФӯЧУФЛ¶ҜКұјдЈ»FКЗЧчУГФЪФӯЧУЙПөДБҰЈ»mКЗПа¶ФФӯЧУЦКБҝЎЈ
ёГјЖЛг№эіМУЙТФПВІҪЦиөГөҪЎЈ
ІҪЦи1Јәёш¶ЁЛщУРФӯЧУөДіхКјО»ЦГЈ»
ІҪЦи2Јәёш¶ЁіхКјөДЛЩ¶ИЈ»
ІҪЦи3Јәёш¶ЁТ»ёцј«РЎөДКұјдФцБҝ Ј¬НЁ№эКҪ(2)јЖЛгФЪ
Ј¬НЁ№эКҪ(2)јЖЛгФЪ КұҝМФӯЧУөДО»ЦГәНЛЩ¶ИЈ»
КұҝМФӯЧУөДО»ЦГәНЛЩ¶ИЈ»
ІҪЦи4Јә·ө»ШІҪЦи2Ј¬јЖЛгПВТ»ҙОөДФӯЧУО»ЦГЎЈ
3 Ҫб№ыУлМЦВЫ
ҫӯ№эід·ЦіЪФҘәуЈ¬ФЪЛЩ¶ИјУФШөДЧчУГПВЈ¬БСОЖіцПЦІўҝӘКјА©Х№ЎЈ¶ФУЪФЛРРҪб№ыІЙУГҝЙКУ»ҜИнјюҪшРРәуҙҰАнЈ¬№ЫІмФӯЧУФЛ¶Ҝ№мјЈЈ¬»жЦЖИзНј2ЛщКҫМШКвКұҝММеПөөДБСОЖА©Х№НјЎЈ
Нј2(a)ЛщКҫОӘ8ЎБ103ІҪөДЛІКұФӯЧУНјЎЈУЙНј2(a)ҝЙјыЈ¬БСОЖТСА©Х№ЦБУТұЯЗшУтЈ¬ҝЙТФҝҙөҪБСОЖөДҝӘҝЪФҪАҙФҪҙуЈ¬БСОЖөДјв¶ЛҪҘҪҘіКПЦОЮРтөДПЦПуЈ¬ҙУХвТ»КұҝМЖрЈ¬БСОЖјв¶ЛөДФӯЧУЦШРВЕЕБжѻэЈ¬ВэВэЖ«АлАнПлҫ§ёсО»ЦГЈ¬Тт¶шІ»Н¬УЪіхКјФӯЧУ№№РНЈ¬ФЪБСОЖјв¶ЛіцПЦ»ыұдЎЈҫ§МеөДХыМеРФФвөҪЖЖ»өЈ¬ДЈРНіКПЦ·Зҫ§ЧҙМ¬Ј¬ФӯЧУЕЕБРөД»мВТөјЦВҫ§МеөДЛЬРФұдРОЎЈ
Нј2(b)ЛщКҫОӘМеПөФЛРРөҪ1ЎБ104ІҪКұ¶ФУҰөДЛІКұФӯЧУНјЎЈҙУНјЦРУТұЯЗшУтҝЙТФәЬЗеіюөШ№ЫІмөҪҝЧ¶ҙөДіцПЦЎЈ°ҙХХҝЧ¶ҙРОәЛ»ъЦЖЈ¬Т»ёцКЗУҰБҰі¬№эБЛФӯЧУјдөДҪбәПБҰЈ¬ФӯЧУјь¶ПБС¶шРОіЙҝЧ¶ҙЈ¬БнТ»ёцКЗҝХО»өДҙуБҝҫЫјҜөјЦВҝЧ¶ҙЎЈПФИ»Ј¬ХвКЗТтОӘЛЬРФұдРОөДҪшТ»ІҪФцЗҝЈ¬ёцұрФӯЧУЦ®јдөДјдП¶ФцҙуЈ¬ЦрІҪ·ўХ№іЙОӘҝХО»Ј¬ҝХО»өДІ»¶П·ўХ№Ј¬ЧоЦХөјЦВ·ЗіЈГчПФөДҝЧ¶ҙіцПЦЎЈТтҙЛЈ¬ҝЙТФНЖ¶ПФӯЧУөД»мВТЕЕБРөјЦВҝЧ¶ҙөДіцПЦЎЈЛжЧЕМеПөөДҪшТ»ІҪФЛРРЈ¬БСОЖјв¶ЛіцПЦёьҙуЗшУтөДФӯЧУ»мВТЕЕБРЈ¬ІъЙъҫЦІҝУҰБҰјҜЦРЎЈХвКұБСОЖТӘјМРшА©Х№РиТӘҝЛ·юёьҙуөДЧиБҰЎЈҙЛНвЈ¬БСОЖјв¶ЛіцПЦБЛ¶Ы»ҜЈ¬БСОЖјв¶Л¶Ы»ҜәуЈ¬ҙЩК№БСОЖХЕҝӘЈ¬БСОЖұдөГФҪАҙФҪҝнЎЈН¬КұТІҪ«ҪөөНјУФШЧчУГП¶ѻэФЪБСОЖјв¶ЛҙҰөДҫЦІҝјҜЦРУҰБҰЈ¬К№өГБСОЖјв¶ЛҙҰөДҫЦІҝјҜЦРУҰБҰЦШРВ·ЦІјЎЈ
Нј2(c)ЛщКҫОӘМеПөФЛРРөҪ2.5ЎБ104ІҪКұөДЛІКұФӯЧУНјЎЈҙЛКұЦШРВ·ЦІјөДҫЦІҝЧоҙуУҰБҰөД·ҪПтҪ«іцПЦРВөДЧУБСОЖЈ¬ҙУФӯАҙөДБСОЖЖҪГжФҫЗЁөҪРВөДБСОЖЖҪГжЈ¬ҙУ¶шЦрҪҘ°ЪНСДёБСОЖЈ¬РОіЙРВөДБСОЖјв¶ЛЎЈІўЗТПтЧЕјҜЦРУҰБҰРЎөДЗшУтА©Х№ЎЈ

Нј2 І»Н¬КұјдІҪМеПөөДБСОЖА©Х№Нј
Fig. 2 Crack propagation maps of system at different time steps
Нј2(d)ЛщКҫОӘМеПөФЪ4.9ЎБ104ІҪКұөДЛІКұФӯЧУНјЎЈУЙНј2(d)ҝЙјыЈ¬МеПөЧЬМе·ЦОӘЙПЎўПВБҪІҝ·ЦЈ¬ҝЙТФҝҙөҪОў№ЫБСОЖөДА©Х№АаЛЖУЪәк№ЫБСОЖЈ¬МеПөТСҫӯұ»АӯҝӘЎЈ
ОӘБЛҪшТ»ІҪЛөГчМеПөБСОЖөДА©Х№ЗйҝцЈ¬МбИЎМеПөЧЬДЬБҝЎЈМеПөЧЬДЬБҝСЭұдНјИзНј3ЛщКҫЎЈҝӘКјҪЧ¶ОЈ¬МеПөөДДЬБҝІ»¶ПөШЙПЙэЈ¬МеПөІ»¶ПөШОьКХДЬБҝЈ¬ХвКЗТтОӘЛжЧЕЛЩ¶ИјУФШЈ¬ФӯЧУөД¶ҜДЬФцјУЈ¬МеПөөДОВ¶ИТІУРЙэёЯЈ¬Тт¶шјУҫзБЛФӯЧУөДФЛ¶ҜЎЈҙуФјФЪ1ЎБ104ІҪКұЈ¬ДЬБҝУРЛщПВҪөЈ¬Т»ЦұөҪ2.5ЎБ104ІҪКұЈ¬ХвТ»№эіМХыёцМеПөөДДЬБҝО¬іЦФЪТ»ёцОИ¶ЁөДЛ®ЖҪЎЈХвТ»ҪЧ¶ОөДМШХчКЗФӯЧУЕЕБРіцПЦ»мВТЈ¬ІъЙъҝХО»Ј¬ҝХО»І»¶ПЗЁТЖЈ¬ҝЧ¶ҙІ»¶ПОьКХҝХО»ІўЗТіӨҙуЈ¬Т»Р©ФӯЧУјдҫаАлФцҙуЈ¬ҪУҪьҪШ¶П°лҫ¶Ј¬ТэЖрПөНіЧЬКЖДЬјхРЎЈ¬ФӯЧУјдөДОьТэБҰұдРЎЎЈҙУ2.5ЎБ104ІҪөҪ3.0ЎБ104ІҪЈ¬ДЬБҝПИЙэәуҪөЈ¬іцПЦТ»ёцРЎёЯ·еЦөЎЈХвТ»ҪЧ¶ОЦчТӘКЗТтОӘБСОЖТӘјМРшА©Х№РиТӘҝЛ·юҫЦІҝУҰБҰјҜЦРЈ¬ЛщТФРиТӘҪшТ»ІҪОьКХДЬБҝЈ¬өұОьКХөДДЬБҝөҪҙпТ»¶ЁіМ¶ИКұЈ¬МеПөөДДЬБҝЧгТФҝЛ·юұЪАЭЈ¬БСОЖјМРшА©Х№Ј¬УЦКН·ЕҙуБҝөДКЖДЬЎЈХвТ»ҪЧ¶О¶ФУҰөДКЗЧУБСОЖөДІъЙъәНЧУБСОЖУлҝЧ¶ҙөД»гјҜ№эіМЎЈЦ®әуөДДЬБҝСЭұд№эіМәНЗ°Т»ҪЧ¶ОөДПаЛЖЈ¬ЛжЧЕМеПөөДҪшТ»ІҪФЛРРЈ¬БСОЖөДҝӘҝЪФҪАҙФҪҙуЈ¬ФӯЧУЦ®јдөДКЖДЬЧчУГФҪАҙФҪИхЈ¬Тт¶шөјЦВХыёцМеПөөДДЬБҝУРТ»ёцЦрІҪөДОўРЎПВҪө№эіМЎЈҙуФјФЪ4.9ЎБ104ІҪКұЈ¬ҝЙТФИПОӘДЬБҝТСҫӯЗчУЪЖҪәвЈ¬ХвКұМеПөТСҫӯұ»АӯҝӘЎЈ
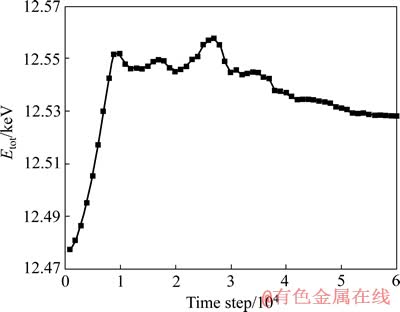
Нј3 МеПөөДЧЬДЬБҝСЭұдНј
Fig. 3 Total energy evolution diagram of system
4 У°ПмБСОЖА©Х№РРОӘөДЦчТӘТтЛШ
4.1 јУФШЛЩВК¶ФБСОЖА©Х№РРОӘөДУ°Пм
ОӘБЛ·ҪұгУліхКјДЈРНЛщЙи¶ЁөДјУФШЛЩВКv=30 m/sПаұИҪПЈ¬·ЦұрСШYХэ·ҪПтК©јУv=20Ўў40әН60 m/sөДЛЩ¶ИјУФШЈ¬ЦұөҪМеПөНкИ«АӯҝӘЈ¬ЖдЛыІОКэЙиЦГұЈіЦІ»ұдЎЈНј4ЛщКҫОӘІ»Н¬јУФШЛЩВКПВМеПөөДДЬБҝСЭұдНјЎЈ
ҙУНј4ЗъПЯaЦРҝЙТФҝҙіцЈ¬МеПөөДДЬБҝТІіКПИФцјУәуПВҪөөДЗчКЖЈ¬ЗъПЯЦРЦ»УРТ»ёцДЬБҝёЯ·еЦөЈ¬ІўЗТіцПЦөДКұјдТӘұИЗъПЯbЦРөДНнЈ¬ҙЛәуТ»ЦұұЈіЦТ»ёцұИҪПОИ¶ЁөДДЬБҝІЁ¶Ҝ·¶О§Ј¬ТтОӘҙЛКұөДјУФШЛЩВКұИҪПРЎЈ¬ЛщТФДЬБҝөДЧоёЯЦөұИЖдЛыЛЩВКПВөДДЬБҝЧоёЯЦө¶јТӘөНЎЈФЪФӯЧУФЛ¶Ҝ№мјЈНјЦРІўОҙ·ўПЦГчПФөДЧУБСОЖУлҝЧ¶ҙөД»гјҜ№эіМЈ¬ХвТІҪвКНБЛЗъПЯaЦРІўГ»УРөЪ¶юёцДЬБҝёЯ·еЦөЎЈЗъПЯcөДРОЧҙУлЗъПЯbПаЛЖЈ¬ө«КЗЗъПЯЦРөДБҪёцДЬБҝ·еЦө¶јұИЗъПЯbЦРөДҙ󣬶шЗТіцПЦөДКұјдТӘұИЗъПЯbЦРөДФзЎЈТтОӘЗъПЯcөДјУФШЛЩВКҙуУЪЗъПЯbөДЈ¬Тт¶шЖд¶ҜДЬҪПёЯЈ¬ҝЙТФҝҙөҪЗъПЯcЧЬМеДЬБҝёЯУЪЗъПЯbөДДЬБҝЎЈБнНвЈ¬ФЪФӯЧУФЛ¶Ҝ№мјЈНјЦРТІ·ўПЦЧУБСОЖөДІъЙъәНЧУБСОЖУлҝЧ¶ҙөД»гјҜКұјдФзУЪЗъПЯbЦРіцПЦөДКұјдЎЈЗъПЯdөДРОЧҙТІАаЛЖУЪЗъПЯbәНcЈ¬ө«КЗөЪ¶юёцДЬБҝёЯ·еЦөіцПЦөДКұјдТӘГчПФФзУЪЗъПЯbәНcіцПЦөДКұјдЎЈХвЦчТӘКЗјУФШЛЩВКФцҙ󣬶ҜДЬТІЛжЦ®ҙу·щ¶ИФцҙуЈ¬ФӯЧУФЛ¶ҜөДјӨБТіМ¶ИјұҫзФцјУЈ¬ФЪПаН¬өДКұјдІҪКэДЪЈ¬МеПөФӯЧУөДО»ТЖҙуөГ¶аЎЈҙУЗъПЯdЦРТІҝЙТФҝҙіцЈ¬ҙуФјҙУ3ЎБ104ІҪәуЈ¬МеПөөДДЬБҝҫНҙпөҪТ»ёц·ЗіЈОИ¶ЁөДЦөЈ¬ҪбәПФӯЧУФЛ¶Ҝ№мјЈНјҝЙЦӘЈ¬ҙУХвЦ®әуЈ¬МеПөТСҫӯұ»АӯҝӘЈ¬АӯҝӘөДКұјдТІГчПФФзУЪЗъПЯaЎўbәНcАӯҝӘөДКұјдЎЈ
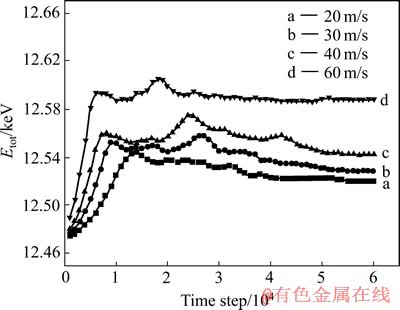
Нј4 І»Н¬јУФШЛЩВКПВМеПөөДЧЬДЬБҝСЭұдЗъПЯ
Fig. 4 Total energy evolution curves of system at different loading rates
4.2 іхКјБСОЖіӨ¶И¶ФБСОЖА©Х№РРОӘөДУ°Пм
ОӘБЛУліхКјДЈРНЛщЙи¶ЁөДБСОЖіӨ¶И10a0ұИҪПЈ¬·ЦұрЙи¶ЁБСОЖіӨ¶И6a0әН15a0Ј¬ЖдЛыМхјюУліхКјДЈРНөДМхјюТ»ЦВЈ¬МбИЎМеПөөДДЬБҝЎЈНј5ЛщКҫОӘІ»Н¬Йи¶ЁБСОЖіӨ¶ИПВМеПөөДДЬБҝСЭұдНјЎЈ
Нј5ЦРЈ¬УЙУЪЖдіхКјјУФШЛЩВКұЈіЦІ»ұдЈ¬№КЗъПЯaЎўbәНcөДҝӘКјЙПЙэҪЧ¶О»щұҫСШЧЕН¬Т»МхЗъПЯСЭұдЎЈЗъПЯaДЬБҝЙПЙэөҪ№ХөгөДЦөГчПФҙуУЪЗъПЯbәНcөДЎЈХвКЗТтОӘЙи¶ЁөДБСОЖіӨ¶ИФҪ¶МЈ¬БСОЖЦ®јдөДФӯЧУКЖДЬФҪөНЈ¬өјЦВКЈПВөДФӯЧУКЖДЬФҪёЯЈ¬ЛщТФМеПөБСОЖөДҪшТ»ІҪА©Х№РиТӘОьКХёь¶аөДДЬБҝАҙҝЛ·юФӯЧУЦ®јдөДКЖДЬЎЈЗъПЯcЦРГ»УРіцПЦҙуөДҝЧ¶ҙҫЫјҜәНіӨҙуЈ¬ХвКЗТтОӘҝЧ¶ҙ»№АҙІ»ј°ҫЫјҜіЙҙуөДҝЧ¶ҙЈ¬БСОЖТСҫӯУлОўРЎҝЧ¶ҙ»гјҜЈ¬ЛщТФІ»ДЬГчПФөШ№ЫІмөҪҝЧ¶ҙіӨҙуөД№эіМЎЈ№КЗъПЯcөДДЬБҝұЈіЦОИ¶ЁөДЛ®ЖҪУЕУЪЗъПЯaәНbөДЎЈҙЛНвЈ¬ЗъПЯa¶ФУҰөДМеПөұ»АӯҝӘөДКұјдІҪКэФЪ5.4ЎБ104ІҪЧуУТЈ¬¶шЗъПЯbөДФјОӘ4.9ЎБ104ІҪЈ¬ЗъПЯcөДФјОӘ4.3ЎБ104ІҪЎЈәЬГчПФЈ¬ХвКЗТтОӘМеПөX·ҪПтөДіӨ¶ИұЈіЦІ»ұдЈ¬БСОЖФҪіӨЈ¬ХыёцМеПөұ»АӯҝӘөДКұјдТІФҪ¶МЎЈ
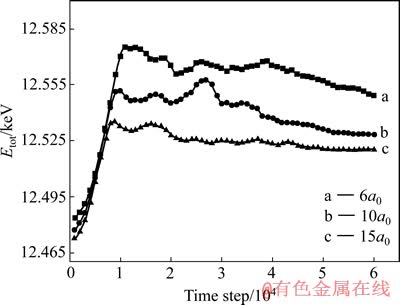
Нј5 І»Н¬БСОЖіӨ¶ИПВМеПөөДЧЬДЬБҝСЭұдЗъПЯ
Fig. 5 Total energy evolution curves of system at different crack lengths
5 ҪбВЫ
1) ҪЁБўБЛөҘҫ§ВБ·ЦЧУ¶ҜБҰС§ДЈДвДЈРНЈ¬НЁ№эМШКвКұҝМөДФӯЧУФЛ¶Ҝ№мјЈНјәНДЬБҝСЭұдНјЈ¬МҪМЦУ°ПмБСОЖА©Х№РРОӘөДЦчТӘТтЛШЎЈ
2) БСОЖјв¶Л¶Ы»ҜәуЈ¬БСОЖјв¶ЛіцПЦОЮРт»мВТөДПЦПуЈ¬ұнПЦОӘБСОЖјв¶ЛіКОЮРтЧҙМ¬Ј¬өјЦВБСОЖјв¶ЛіцПЦ»ыұдЎЈ
3) ЛжЧЕјУФШЛЩВКөДФцҙуЈ¬МеПөөДФӯЧУФЛ¶ҜјУҫзЈ¬МеПөНкИ«ұ»АӯҝӘөДКұјдұд¶МЈ»іхКјБСОЖіӨ¶ИФҪіӨЈ¬ЛщРиТӘОьКХөДДЬБҝФҪЙЩЈ¬ХыёцМеПөұ»АӯҝӘөДКұјдТІФҪ ¶МЈ¬ФӯЧУФЛ¶ҜөДПёҪЪұнПЦФҪІ»ГчПФЎЈ
REFERENCES
[1] STALEY J T, LIU J, HUNT W H Jr. Aluminum alloys for aerostructures[J]. Advanced Materials & Processes, 1997, 10: 17-19.
[2] MACHOVA A, KROUPA F. Atomistic modelling of contribution of dislocations to crack opening displacements[J]. Materials Science and Engineering A, 1997, 234/236: 185-188.
[3] MAKI-JASKARI M, KASKI K, KURONEN A. Simulations of crack initiation in silicon[J]. Computational Materials Science, 2000, 17: 336-342.
[4] ЦЬ№ъ»Ф, ёЯҝЛзв, Нт·ўИЩ, ЗЗАыҪЬ, сТОдСп. ОўБСОЖУъәП№эіМөД·ЦЧУ¶ҜБҰС§ДЈДв[J]. ЧФИ»ҝЖС§ҪшХ№, 2001, 11(3): 300-305.
ZHOU Gou-hui, GAO Ke-wei, WAN Fa-rong, QIAO Li-jie, CHU Wu-yang. Molecular dynamics simulation of micro crack healing process[J]. Progress in Natural Science, 2001, 11(3): 300-305.
[5] WEI D B, HAN J T, TIEU K, JIANG Z Y. Simulation of crack healing in BCC Fe[J]. Scripta Materialia, 2004, 51(6): 583-587.
[6] НхПюҫк, ЦмұҰИ«, НхәмГ·. ОВ¶И¶ФөҘҫ§ВББСОЖА©Х№У°ПмөД·ЦЧУ¶ҜБҰС§ДЈДв[J]. ПөНі·ВХжС§ұЁ, 2010, 22(2): 534-536.
WANG Xiao-juan, ZHU Bao-quan, WANG Hong-mei. Effects of temperature on growth of monocrystalline aluminum crack: A molecular dynamics study[J]. Journal of System Simulation, 2010, 22(2): 534-536.
[7] WU Wen-ping, YAO Zong-zhuan. Molecular dynamics simulation of stress distribution and microstructure evolution ahead of a growing crack in single crystal nickel[J]. Theoretical and Applied Fracture Mechanics, 2012, 62: 67-75.
[8] өҘөВұт, Ф¬ БЦ, №щ ұу. өҘҫ§НӯНдЗъБСОЖГИЙъәНА©Х№өД·ЦЧУ¶ҜБҰС§ДЈДв[J]. №ю¶ыұх№ӨТөҙуѧѧұЁ, 2003, 35(10): 1183-1185.
SHAN De-bin, YUAN Lin, GUO Bin. Molecular dynamics simulation of bending crack initiation and growth of single crystal Cu[J]. Journal of Harbin Institute of Technology, 2003, 35(10): 1183-1185.
[9] №щСЕ·ј, ёЯЛчОД. ·ЦЧУ¶ҜБҰС§ДЈДвБСОЖА©Х№ј°Па№ШіЯҙзРРОӘ[J]. ұұҫ©Ҫ»НЁҙуѧѧұЁ, 2005, 29(4): 5-9.
GUO Ya-fang, GAO Suo-wen. Atomistic simulation of crack propagation and its size-dependent behavior[J]. Journal of Beijing Jiaotong University, 2005, 29(4): 5-9.
[10] CAO A-jing, WEI Yue-guang. Atomistic simulations of crack nucleation and intergranular fracture in bulk nanocrystalline nickel[J]. Physical Review B, 2007, 76: 024113-5.
[11] ФшПй№ъ, РнКйЙъ, іВ»ӘСа. ГҫәПҪрБСОЖ¶Ҙ¶ЛЛЬРФұдРОәНК§Р§»ъАнөД·ЦЧУ¶ҜБҰС§ДЈДв[J]. ЛДҙЁҙуѧѧұЁ, 2011, 48(1): 173-178.
ZENG Xiang-guo, XU Shu-sheng, CHEN Hua-yan. Molecular dynamics simulation on plastic deformation and failure mechanisms around a crack tip for magnesium alloys[J]. Journal of Sichuan University, 2011, 48(1): 173-178.
[12] ZHANG Y F, LIU X Y, MILLETT P C, TONKS M, ANDERSSON D A, BINER B. Crack tip plasticity in single crystal UO2: Atomistic simulations[J]. Journal of Nuclear Materials, 2012, 430(1/3): 96-105.
[13] ХФСЮәм, АоУўҝҘ, СоЦҫ°І, ХЕ№гІЖ. ҙшҝЧ¶ҙөДҪрКфАӯЙмөД·ЦЧУ¶ҜБҰС§[J]. јЖЛгОпАн, 2006, 23(3): 343-349.
ZHAO Yan-hong, LI Ying-jun, YANG Zhi-an, ZHANG Guang-cai. Molecular dynamics simulation of Cu with a hole under minus static pressures[J]. Chinese Journal of Computational Physics, 2006, 23(3): 343-349.
[14] ЦмЦҫРЫ, ХЕ әи, Бхі¬·е, ЖлОАәк, ТЧөӨЗа, АоЦҫіЙ. Ni-AlәПҪрДэ№М№эіМөД·ЦЧУ¶ҜБҰС§ДЈДв[J]. ЦР№ъУРЙ«ҪрКфС§ұЁ, 2009, 19(8): 1409-1416.
ZHU Zhi-xiong, ZHANG Hong, LIU Chao-feng, QI Wei-hong, YI Dan-qing, LI Zhi-cheng. Molecular dynamics simulation for solidification process of Ni-Al alloys[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(8): 1409-1416.
[15] ЗсҝЛЗҝ, Ач әз, Лп ҫ§, УИҝЎ»Ә, ИОУўАЪ, АоЗм·б. Cu66Ti34·Зҫ§әПҪрДэ№М№эіМөД·ЦЧУ¶ҜБҰС§ДЈДв[J]. ЦР№ъУРЙ«ҪрКфС§ұЁ, 2011, 21(9): 2151-2156.
QIU Ke-qiang, LI Hong, SUN Jing, YOU Jun-hua, REN Ying-lei, LI Qing-feng. Solidification process of Cu66Ti34 amorphous alloy simulated by molecular dynamics[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(9): 2151-2156.
[16] GUO Yong-bo, LIANG Ying-chun. Atomistic simulation of thermal effects and defect structures during nanomachining of copper[J]. Transactions of Nonferrous Metals Society of China, 2012, 22(11): 2762-2770.
[17] іВ Гч, Ао ёп, ХЕОД·Й. ДЙГЧіЯ¶ИҝЧ¶ҙЦЬО§УҰБҰјҜЦРПЦПу·ЦОц[J]. ІДБПөјұЁB, 2011, 25(3): 131-134.
CHEN Ming, LI Ge, ZHANG Wen-fei. Discussion on the phenomenon of stress concentration around hole in nano-scale[J]. Materials Review B, 2011, 25(3): 131-134.
[18] KARIMI M, ROARTY T, KAPLAN T. Molecular dynamics simulations of crack propagation in Ni with defects[J]. Modelling Simul Mater Sci Eng, 2006, 14: 1409-1420.
[19] TANG T, KIM S H, HORSTEMEYER M F, WANG P. Atomistic modeling of crack growth in magnesium single crystal[J]. Engineering Fracture Mechanics, 2011, 78: 191-201.
[20] TERENTYEV D, ZHURKIN E E, BONNY G. Emission of full and partial dislocations from a crack in BCC and FCC metals: An atomistic study[J]. Computational Materials Science, 2012, 55: 313-321.
[21] ACKLAND G J, VITEK V. Many-body potentials and atomic-scale relaxations in noble-metal alloys[J]. Physical Review B, 1990, 41(15): 10324-10333.
(ұајӯ іВОАЖј)
»щҪрПоДҝЈә№ъјТЧФИ»ҝЖС§»щҪрЧКЦъПоДҝ(11362017)Ј»ҪӯОчКЎЧФИ»ҝЖС§»щҪрЧКЦъПоДҝ(2010GQC0803)Ј»ҪӯОчКЎҪМУэМьҝЖјјВдөШјЖ»®ҝЖС§З°СШПоДҝ(KJLD12073)
КХёеИХЖЪЈә2013-08-29Ј»РЮ¶©ИХЖЪЈә2014-03-12
НЁРЕЧчХЯЈәБхПюІЁЈ¬ҪМКЪЈ¬І©КҝЈ»өз»°Јә0791-83953108Ј»E-mail: liuxb2000@sina.com

