������ʱ��: 2013-12-27 09:56
ϡ�н��� 2014,38(06),1106-1113 DOI:10.13373/j.cnki.cjrm.2014.06.027
���ٿ���өʯ������ʯ���ӽṹ�ĵ�һ��ԭ���о�
��Ӣ ��ع�� ������ ������ ������
ʡ������������ɫ������Դ������ù����ص�ʵ����
����������ѧ������Դ����ѧԺ����ӹ�����ϵ
���ϴ�ѧ��Դ�ӹ������﹤��ѧԺ����ӹ�����ϵ
���ϿƼ���ѧ��������ԴѧԺ����ӹ�����ϵ
ժ Ҫ��
���û����ܶȷ������۵ĵ�һ��ԭ��, ������������ٿ�өʯ�ͷ���ʯ�ĵ��ӽṹ, ��ˮ���Ӻ;۱�ϩ������3�ֿ������������������, �����õ����︡ѡ������֤��ˮ��ϵ�о۱�ϩ������3�ֿ������������ܵĹ�ϵ������������:���ٿ�өʯ�ͷ���ʯ���Ż������þ����ܶȷ������� (LDA) �µ�CA-PZ������غ���, �ض��ֱܷ�Ϊ277, 270��275 e V;�ܴ��ṹ����3�ֿ�������ھ�Ե��, ��Caԭ��̬�ܶ���ɺ�����, ��˻�ѧ���Էdz�����, �ڸ�ѡ�����б��ֳ����Ƶĸ�ѡ����;Mulliken���ӷ�������3�ֿ���Caԭ��������ɴ�С˳��Ϊ����ʯ>���ٿ�>өʯ���ڿ��о۱�ϩ�����ſ���������ˮ��������������, �������ܾ�Ϊ��ֵ, ��С˳��Ϊ���ٿ�>����ʯ>өʯ, ˵����Ȼp H (δ����p H������) �����¾۱�ϩ���ƶ�3�ֿ�������������, ����ǿ��Ϊөʯ>����ʯ>���ٿ����︡ѡ����������, �۱�ϩ����Ϊ���Ƽ�ʱ, ����Ȼp H������, ���ٿ�өʯ�ͷ���ʯ�Ļ����ʷֱ�Ϊ76.03%, 18.59%��33.68%, ��ʱ�۱�ϩ���Ƶ�����ǿ��˳��Ϊөʯ>����ʯ>���ٿ�, �����ģ���������Ϊ��һ���˽���ٿ�өʯ�ͷ���ʯ�ɸ��ԵIJ��켰���ƿ��︡ѡ��ҩ�������ṩ���۲ο���
�ؼ��ʣ�
���ٿ�;өʯ;����ʯ;��һ��ԭ��;��ѡ;
��ͼ����ţ� TD923;TD97
����飺��Ӣ (1984-) , Ů, �Ĵ��ڽ���, ��ʿ, ��ʦ, �о�����:��ѡ�����빤��;E-mail:zhyingcsu@163.com;;��ع��, ����;�绰:0731-88830545;E-mail:wangyh@mail.csu.edu.cn;
�ո����ڣ�2013-11-01
��������ʮ����Ƽ�֧�żƻ���Ŀ (2012BAB10B05);����������ѧУ�˲�������Ŀ (ʡ��) (KKSY201321123);����������ѧ�������Ի��� (20140910) ����;
First-Principle Theory Calculation of Electronic Structures of Scheelite, Fluorite and Calcite
Zhang Ying Wang Yuhua Hu Yuehua Wen Shuming Wang Jinming
State Key Laboratory of Complex Nonferrous Metal Resources Clean Utilization
Department of Mineral Processing Engineering, Faculty of Land Resource Engineering, Kunming University of Science and Technology
Department of Mineral Processing Engineering, School of Minerals Processing and Bio-Engineering, Central South University
Department of Mineral Processing Engineering, School of Environment and Resource, Southwest University of Science and Technology
Abstract��
The electronic structures of scheelite, fluorite and calcite and the adsorption energy of H2 O and PA-Na adsorbing on the three kinds of mineral surface were calculated using first-principle method based on the density functional theory ( DFT) . Single mineral flotation tests verified the adsorption energy relationship of PA-Na attached to minerals in the water system. The calculation results showed that exchange / correlation function ( CA-PA) of local-density approximation ( LDA) was used to optimize electronic structures of scheelite, fluorite and calcite, and cut-off energies were 277, 270 and 275 e V, respectively. Energy band structures showed that the three minerals were all insulators and their densities of Ca were similar extremely. Their chemical activities were very similar, so that the minerals showed similar flotation performance. The Mulliken population analysis showed that the charge of Ca of the three mineralswas as follows: calcite > scheelite > fluorite. Sodium polyacrylate ( PA-Na) was absorbed on mineral surfaces when H2 O molecules were released in the pulp, and the adsorption energies were negative in the order of scheelite > calcite > fluorite. This showed that PANa could inhibit the three minerals in the order of fluorite > calcite > scheelite at the nature p H ( p H adjusting agent was not added) .Single mineral flotation test results showed that as an inhibitor of PA-Na, the recoveries of scheelite, fluorite and calcite were 76. 03%, 18. 59% and 33. 68%, respectively, at the nature p H. The inhibiting ability of PA-Na was in the order of fluorite > calcite > scheelite, which was consistent with the calculation results. This work provided a theoretical reference for the research on flotation performances of calcite, scheelite and fluorite and the development reagents.
Keyword��
scheelite; fluorite; calcite; first-principle theory; flotation;
Received�� 2013-11-01
���Ǹ��۵��ϡ�н���[1], ��һ����Ҫ��ս����Դ�����ٿ�����Ҫ�к��ٿ��ٿ�ͺڰ���ٿ�, ��ѡ�ĺ��ٿ��ѻ����ݽ�, �����ڰ��ٿ����ϸ��Ƕ���Ŀ�ʯ, ���ڲ��ø�ѡ���з������, ���ѡ��Ƽ����������ⷽ���ѽ����˴������о�[2,3,4,5]�����ٿ���������������ɸ������Ƶĺ�����ʯ����, ��өʯ�ͷ���ʯ��, �Ӷ�ʹ�ð��ٿ���өʯ������ʯ���ڷ���[6,7,8,9]��
����ĸ�ѡ��Ϊȡ���ڿ��������, �����������ȡ���ڿ���ĵ��ӽṹ, ��˴Ӱ��ٿ�өʯ�ͷ���ʯ3�ֿ�����ӽṹ�����ҵ����ǵ����ڲ���, ��ָ����ѡʵ�����Ե÷dz���Ҫ�����������ż���������Լ����ӻ�ѧ���۵ķ�չ, ͨ�������ģ����ᄃ��ṹ������ҩ���������, �Ӷ���ʾ��ʯ������Ŀ�Ŀ���ṹ�IJ����Լ���ҩ��������û���, ��ָ����ѡʵ����Ϊ����ʵ, �����ܶȷ����ĵ�һ��ԭ�����о����ᄃ��ṹ���������ʵ���������[10,11], ����ѡ�����о���Ӧ�ñȽϹ㷺[12,13,14,15,16], �����õ�һ��ԭ���о����ٿ�өʯ�ͷ���ʯ��ѡ���ʲ���ı������١�
���IJ����ܶȷ�����һԭ���о��˰��ٿ�өʯ�ͷ���ʯ���弰������ӽṹ����, ��ģ����ˮ��ϵ�о۱�ϩ������3�ֿ������������Ϊ�IJ���, �о�������ڴӱ���������3�ֿ���Ŀɸ��Բ��������Ҫ������, �Ӷ�Ϊ�о����ٿ���өʯ������ʯ��ѡ�����ṩ���۲ο���
1���㷽����ģ��
�����ܶȷ����ĵ�һ��ԭ������, ��������Material Studio 4.4�е�CASTEPģ��, ���ٿ�өʯ�ͷ���ʯ3�ֺ��ƿ�����ܴ��ṹ������̬�ܶȺ�Mulliken���ӽ��м��㡣Ϊ��ʹ���㾧�����ͽ������Ƚӽ�ʵ��ֵ, ���ȶ�3�ֺ��ƿ����ԭ��ģ�ͽ����Ż�����, ��ѡȡ�ϼѵĽ�������������ƽ�沨�ض��ܡ�������ѡȡ����������������ƽ�沨�ض��ܺ���ѡȡ�ĺ�������Ӧ��Ĭ��ֵ, 3�ֿ����ԭ��ģ����ͼ1��ʾ��ͨ���Լ��ֺ����������ĶԱ�, �����ܶȷ������� (LDA) �µ�CA-PZ������������������ľ�������ʵ��ֵ��Ϊ�ӽ�, ���3�ֺ��ƿ���Ľ�����������������LDA�µ�CA-PZ���������������ض��ܵļ��������1��ʾ��
��1��ƽ�沨�ض��ܲ��Ա���:���ٿ�Ľض���Ϊ277 e Vʱ��Ϊ����, �侧������ļ���ֵa=b=0.52372 nm��ʵ��ֵa=b=0.52429 nm[17]�����Ϊ0.1%;өʯ�Ľض���Ϊ270 e Vʱ, �侧������ļ���ֵa=b=c=0.54624 nm��ʵ��ֵa=b=c=0.54631 nm[18]������Ϊ0.01%;����ʯ�Ľض���Ϊ275 e Vʱ, ģ�����ó��ľ������ֵΪa=b=0.49868 nm, ��ʵ��ֵa=b=0.4988 nm[19]������Ϊ0.02%��3�ֿ���ļ�������ʵ������������С, �������������õķ����Լ�ѡȡ�IJ����ǿɿ��ġ�
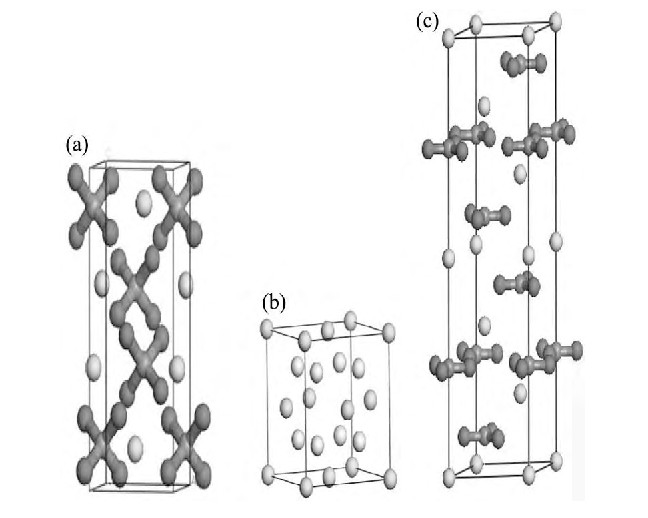
ͼ1 ���ٿ�, өʯ�ͷ���ʯ�ĵ���ģ��Fig.1 Unit cell models of scheelite (a) , fluorite (b) and cal-cite (c)
��1 ��������ΪLDA-CA-PZʱ, �ض��ܲ��Խ��Table 1 Results of cutoff energy testing by LDA-CA-PZ ����ԭͼ

Note:��= (Experimental data-Lattice constant) /Experimental data��100%
��1 ��������ΪLDA-CA-PZʱ, �ض��ܲ��Խ��Table 1 Results of cutoff energy testing by LDA-CA-PZ
2���������
2.1�ܴ��ṹ����
���ٿ�өʯ�ͷ���ʯ���ܴ��ṹ��ͼ2��ʾ, ȡ�����ܼ� (EF) ��Ϊ������㡣����������:���ٿ�Ľ�������Ϊ4.184 e V, ��Minoru Itoh�ȼ������4.25 e V�ȽϽӽ�[20,21];өʯ�Ľ�������Ϊ6.902 e V;����ʯ�Ľ�������Ϊ4.835 e V, ��Andrew J Skinner�ȼ������ (4.4��0.2) e V�ȽϽӽ�[22]��ģ�������Ľ������ȸ��ڻ����ʵ��ֵ��Ҫ������GGA�����µ�DFT, �Ե��������֮��Ľ����������ô������������[23]�����ڰ뵼��Ľ�������һ����2 e V����, ��Ե��Ľ������������[24], ������ܴ��ṹ��������֪, ���ٿ�өʯ�ͷ���ʯ�����ھ�Ե�塣

ͼ2 ���ٿ�, өʯ�ͷ���ʯ���ܴ��ṹFig.2 Band structures of scheelite (a) , fluorite (b) and cal-cite (c)
2.2̬�ܶȷ���
���ٿ�өʯ�ͷ���ʯ3�ֿ����̬�ܶ���ͼ3��ʾ����ͼ3���Կ���, ���ٿ���ʯ�����ܼ�������̬�ܶ���Ҫ������2p�������, ��өʯ��Ҫ�ɷ���2p������ɡ����ٿ���ʯ��өʯ������Ca��̬�ܶ���ɷdz�����, 3�ֿ�����-40 e V������̬�ܶ���Ҫ��Ca3s������, ������-20 e V������̬�ܶ���Ҫ��Ca3p������ס����ʹ�ÿ������Ca���ﻯ��������, ����ڸ�ѡ�����п�������Ca���ֳ����ƵĻ�ѧ����, ���Է��롣
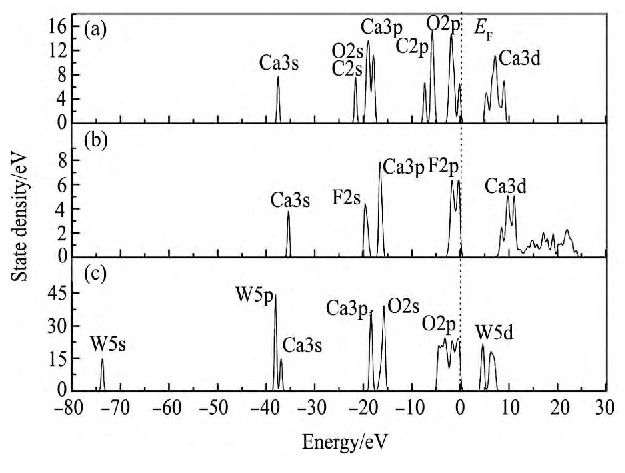
ͼ3 3�ֿ����̬�ܶȱȽ�Fig.3 Density of state of three minerals (a) Calcite; (b) Fluorite; (c) Scheelite
3�ֿ���̬�ܶȵ�С�����Ҫ����: (1) ��-75 e V�������ٿ�W5s�������̬�ܶ���С�Ĺ���, ���������ֿ�����û��, ������������λ�õ�����̫��, һ����Ϊ�����Բμӻ�ѧ��Ӧ; (2) ���ٿ�ͷ���ʯ�����ܼ�������̬�ܶ���Ҫ������2p�������, өʯ�����ܼ������ļ۴���Ҫ��F2p������, ���ڴ��ڷ����ܼ���������̬��ɾ��нϸߵĻ�ѧ����, ���, ���ٿ�ͷ���ʯ�ڲ��뻯ѧ��Ӧʱ��O�Ļ��Խ�ǿ, ��өʯ����F�Ļ��Խ�ǿ; (3) өʯ�ͷ���ʯ�ĵ����ײ���̬�ܶ���Ҫ��Ca3d�������, �����ٿ��ײ�̬�ܶ���Ҫ��W5d������ס�������Щ3�ֿ���̬�ܶȵIJ����п��ܳ�Ϊ����ѡ���Է�������ݡ�
2.3 Mulliken���ӷ���
Mulliken�ص�������Mulliken�����һ�ֱ�ʾ����ڸ����ԭ��֮��ֲ�����ķ�����ͨ��Mulliken���ӷ������Կ���ģ����ϵ�ĵ�ɷֲ���ת�ƺ����γɵļ������ʵ����[25]��
���ٿ��Oԭ�ӡ�Caԭ�Ӻ�Wԭ�����Ż�ǰ�ļ۵��ӹ���ΪO2s22p4, Ca3s23p64s2, W5s25p65d4, �Ż���ԭ�ӵ�Milliken����ֵ���2��ʾ���ӱ�2�п�֪:���ٿ��Ż���ļ۵��ӹ���ΪO2s1.862p4.84, Ca3s2.133p6.003d0.53, W5s2.305p6.545d3.71��Caԭ�Ӻ�Wԭ��Ϊ���ӹ���, ��ԭ���������Ϊ+1.35 e, ��ԭ���������Ϊ+1.46 e��Oԭ��Ϊ��������, �������Ϊ-0.70 e��
өʯ��Fԭ�Ӻ�Caԭ�����Ż�ǰ�ļ۵��ӹ���ΪF2s22p5, Ca3s23p64s2, �Ż���ļ۵��ӹ���ΪF2s1.962p5.71, Ca3s2.163p6.004d0.51, Caԭ���������Ϊ+1.32 e, Ϊ���ӹ���, Fԭ���������Ϊ-0.66 e, Ϊ�������塣
��2 ���ٿ�өʯ�ͷ���ʯԭ�ӵ�Mulliken���ӷ���Table 2 Mulliken atomic population analysis of scheelite, fluorite and calcite ����ԭͼ
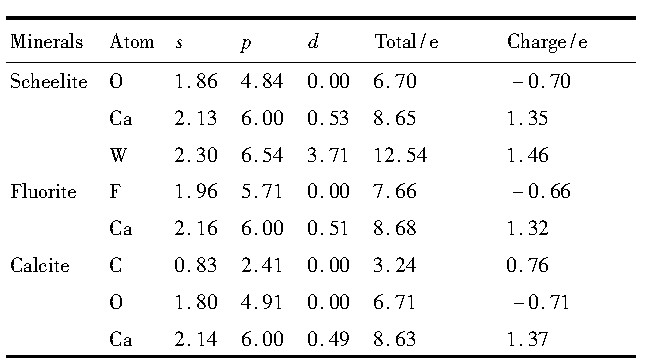
��2 ���ٿ�өʯ�ͷ���ʯԭ�ӵ�Mulliken���ӷ���Table 2 Mulliken atomic population analysis of scheelite, fluorite and calcite
����ʯ��Cԭ�ӡ�Oԭ�Ӻ�Caԭ�����Ż�ǰ�ļ۵��ӹ���ΪC2s22p2, O2s22p4, Ca3s23p64s2, �Ż���ļ۵��ӹ���ΪC2s0.832p2.41, O2s1.802p4.914s0.42, Ca3s2.143p6.003d0.49, Caԭ�Ӻ�Cԭ��Ϊ���ӹ���, ��ԭ���������Ϊ+1.37 e, ̼ԭ���������Ϊ+0.76 e, Oԭ��Ϊ��������, �������Ϊ-0.71 e��
�ۺϱ�2�����ݷ�����֪:���ٿ�өʯ�ͷ���ʯ�еĸ�ԭ�Ӿ�������, ������ɴ�С˳��Ϊ����ʯ>���ٿ�>өʯ;���ٿ�ͷ���ʯ�е���ԭ�Ӵ�����, �ٺ�̼ԭ�Ӵ�����;өʯ�еķ�ԭ�Ӵ����硣
����Mulliken����ֵ�����ֳ����������Ժ����Ե�ǿ��, ����ֵԽ��������Ĺ�����Խǿ, ԽС���������������Խǿ�����ٿ�өʯ�ͷ���ʯ����Mulliken����ֵ���ڱ�3��, �ɱ�3�����ݷ�����֪:���ٿ�ͷ���ʯ�е�O-Ca����өʯ�е�F-F�������ֳ��ϴ��������, �ײ������Ӽ�����;���ٿ��е�O-W���ͷ���ʯ�е�C-O����Ҫ�Թ�����Ϊ��, �����ڴ��ڻ��Žṹ��, ������ĥ����������Զ���, ��˰��ٿ�ͷ���ʯ���������������ҪΪ���Ӽ�����;�Ӽ��IJ���ֵ���Կ���, ���ٿ���Ca-O���ͷ���ʯ��CaO���IJ�������С, ֻ��0.001 nm, �Ӷ����DZ�����Ķ����������, ���ٿ�ͷ���ʯ���涼�мۼ�δ���͵ĸ�����, өʯ�����F-F����������, �������ӵ�ˮ�������ܱȱ�������ӵ�С, ��������ˮ��������Һ, ʹ�ñ���Ҳ�����ۼ�����������, 3�ֿ������Ļ�ѧ���Էdz�����, ���, �ڸ�ѡ�����б��ֳ����Ƶĸ�ѡ���ܡ�
��3 ���ٿ�өʯ�ͷ���ʯ����Mulliken���ӷ���Table 3Mulliken bond population analysis of scheelite, fluorite and calcite ����ԭͼ

��3 ���ٿ�өʯ�ͷ���ʯ����Mulliken���ӷ���Table 3Mulliken bond population analysis of scheelite, fluorite and calcite
2.4�����ܷ���
��4�г�3�ֿ����������Сʱ��ԭ�Ӳ�������ղ���, ����ģ����ͼ4��ʾ�����ٿ� (111) ���溬��6��ԭ�Ӽ�1.2 nm����ղ��ȵı���ṹ�ܹ�������������������, ��ʱ�ı�����Ϊ0.7202 J��m-2;өʯ (111) ���溬��3��ԭ�Ӽ�1.0 nm����ղ��ȵı���ṹ�ܹ�������������������, ������Ϊ0.5517 J��m-2;����ʯ (104) ���溬��21��ԭ�Ӽ�1.2 nm����ղ��ȵı���ṹ�ܹ�������������������, ������Ϊ0.4350 J��m-2��
2.5ˮ��ϵ�о۱�ϩ�����ڿ�������������
�����ܶȷ�������ģ��ˮ���Ӻ;۱�ϩ���Ʒ����ڰ��ٿ� (111) ��өʯ (111) �ͷ���ʯ (104) ���������, ��ˮ���Ӻ;۱�ϩ���Ʒֱ�����3�ֿ�����治ͬλ����, ͨ���Ż����ͺͼ���������, ��������С������λ��Ϊ���ȶ�������ģʽ��������Ϊ����ǰ��������������ı仯, ���С���Ա�ʾ������ϵ���ȶ���[26]��ҩ���ڿ������������ܸ�����ʽ����:
��4 3�ֿ���ı����ܷ���Table 4 Surface energy analysis of minerals ����ԭͼ

��4 3�ֿ���ı����ܷ���Table 4 Surface energy analysis of minerals

ͼ4 3�ֿ���ı��浥��Fig.4 Surface unit cells of minerals
(a) Scheelite (111) ; (b) Fluorite (111) ; (c) Calcite (104)

ʽ��, EadsΪ������ (k J��mol-1) ;E (����+ҩ��) Ϊ��������ϵ�������� (k J��mol-1) ;E����Ϊ������������ (k J��mol-1) ;Eҩ��Ϊҩ�������� (k J��mol-1) ��������Ϊ��, ��������Ϊ���ȷ�Ӧ, �������ֵԽ��, ˵��ҩ���ڱ��������Խǿ, ��������Խ��������
���ھ۱�ϩ���Ʒ�����̫���ڼ�������������, ���ѡȡ���IJ��ֹ����Žṹ��Ԫ����ģ�⡣ˮ���Ӻ;۱�ϩ���Ʒ����ڿ���������ȶ�����ģʽ�������ֱܷ����5, 6��ʾ��
�ɱ�5��6�Ľ�����Կ���, ˮ�ڰ��ٿ�өʯ�ͷ���ʯ���������������, ���۱�ϩ������3�ֿ�������������ǿ, �Ҹ���������
ͼ5��6����, ˮ��������ٿ�����ʱ������ͬһWO3���ŵ�������ԭ���ϵ���������СΪ-225.70 k J��mol-1, ��өʯ����ʱ�����ڲ�ͬ���Ӳ��������ԭ���ϵ���������СΪ-112.03k J��mol-1, �뷽��ʯ����ʱ������ͬһCO3���ŵ�������ԭ���ϵ���������СΪ-110.52 k J��mol-1���۱�ϩ���Ʒ�������ٿ�����ʱ������ͬһ���Ӳ�ĸ�ԭ���ϵ���������СΪ-359.27 k J��mol-1, ������өʯ (111) ��ͬһ���Ӳ�ĸ�ԭ���ϵ���������СΪ-405.32 k J��mol-1, �۱�ϩ���������ڷ���ʯ (104) ��ͬx��ĸ�ԭ���� (CP1) ����������СΪ-289.06 k J��mol-1��ˮ���Ӻ;۱�ϩ���Ʒ�����3�ֿ�������������ģ�ͷֱ���ͼ5, 6��ʾ��
��5 ˮ������3�ֿ�������������Table 5Adsorption energy of H2O adsorbed on mineral surfaces (k J��mol-1) ����ԭͼ

��5 ˮ������3�ֿ�������������Table 5Adsorption energy of H2O adsorbed on mineral surfaces (k J��mol-1)
��6 �۱�ϩ���Ʒ�����3�ֿ�������������Table 6 Adsorption energy of PA-Na adsorbed on mineral surfaces (k J��mol-1) ����ԭͼ
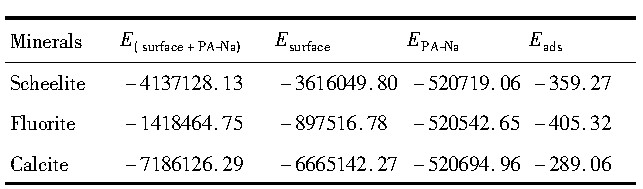
��6 �۱�ϩ���Ʒ�����3�ֿ�������������Table 6 Adsorption energy of PA-Na adsorbed on mineral surfaces (k J��mol-1)
���ھ۱�ϩ������3�ֿ�������ˮ��ϵ�з�������, ����ǰ��ȻҪ�ż�����������Ѿ�������ˮ��Ĥ, ��˾۱�ϩ������ˮ��Һ����3�ֿ����������������С���Զ���Ϊ:


ͼ5 ˮ������3�ֿ�������������ģ��Fig.5 Models of H2O adsorbed on minerals
(a) Scheelite; (b) Fluorite; (c) Calcite
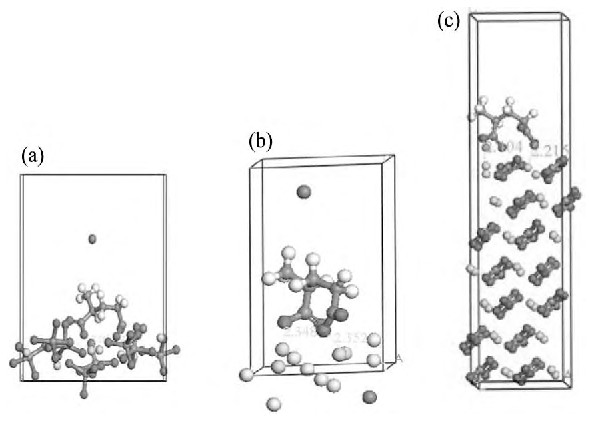
ͼ6 �۱�ϩ���Ʒ�����3�ֿ�������������ģ��Fig.6 Models of PA-Na adsorbed on minerals
(a) Scheelite; (b) Fluorite; (c) Calcite
ʽ��, EaΪ�۱�ϩ�����ڿ�������������;EbΪˮ�����ڿ�������������;��EadsΪ��˵���۱�ϩ���ƿ��Կ˷�ˮ�����ڿ����������������, EadsΪ��������۱�ϩ�������Կ˷�ˮ�����ڿ����������������, EadsԽС�۱�ϩ�����ڿ�����������Խ��������
����ʽ (2) ���������ܵļ���, �������7���Ӽ���������3�ֿ�����ˮ��ϵ����۱�ϩ���Ʒ�����Ӧ�������ܾ�Ϊ��ֵ, �����ֵ��С˳��Ϊөʯ>����ʯ>���ٿ�˵���۱�ϩ������3�ֿ�����������ʹ�����ܵ���������, ��˾۱�ϩ��������Ȼp H (δ����p H������) �����¶�3�ֿ�����������˳��Ϊөʯ>����ʯ>���ٿ�
2.6�����︡ѡ����
Ϊ�˽�һ����֤ģ�������, ���ٿ�өʯ�ͷ���ʯ������Ϊ����, �Ծ۱�ϩ���� (25 mg��L-1) Ϊ���Ƽ���̼����/����Ϊp H��������731 (75mg��L-1) Ϊ���ռ������˵����︡ѡ����, ��p Hֵ��3�ֿ���ɸ��Ե�Ӱ����ͼ7��ʾ��
��7 �۱�ϩ������ˮ��ϵ���ڿ�������������/e V Table 7 Adsorption energy of PA-Na adsorbed on mineral surface in H2O (k J��mol-1) ����ԭͼ
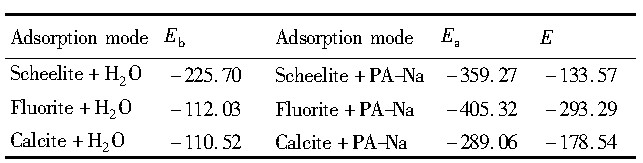
��7 �۱�ϩ������ˮ��ϵ���ڿ�������������/e V Table 7 Adsorption energy of PA-Na adsorbed on mineral surface in H2O (k J��mol-1)
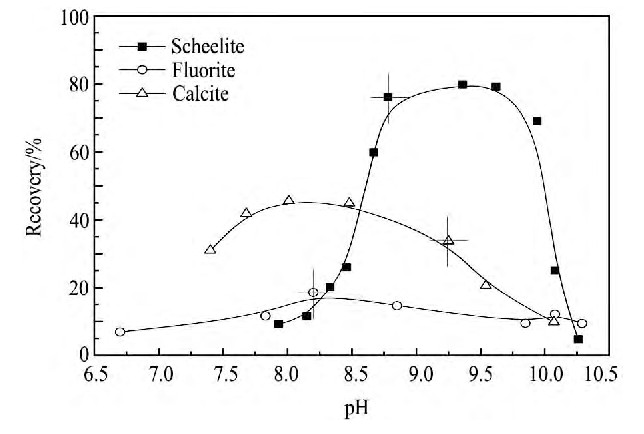
ͼ7 �۱�ϩ����Ϊ���Ƽ���731Ϊ���ռ�ʱ, ��p Hֵ�Ժ��ƿ���ɸ��Ե�Ӱ��Fig.7 Recovery of calcium minerals as a function of p H in presence of PA-Na and 731.c (731) =75 mg��L-1, c (PA-Na) =25 mg��L-1
ͼ7�ĸ�ѡ�������:���þ۱�ϩ���������Ƽ���731Ϊ���ռ�ʱ, ����p Hֵ������, 3�ֿ���Ļ����ʾ������Ӻ�;p H=8.7~10��Χ��, ���ٿ�Ļ��������Ը���өʯ������ʯ�Ļ�����, ��ʱ���ٿ�Ļ����ʾ�������80%����, өʯ�Ļ�������15%~10%֮��, ����ʯ�Ļ����ʵ���40%���Ӷ�����˵��, �۱�ϩ������өʯ�Ͱ��ٿ����Ч���Ƽ�, �п���ʵ�ְ��ٿ���өʯ������ʯ�ĸ�ѡ���롣����Ȼp H (δ����p H������) ������, ���ٿ�өʯ�ͷ���ʯ��ѡʱ��p Hֵ�ֱ�Ϊ8.78, 8.20��9.25, ��Ӧ�Ļ����ʷֱ�Ϊ76.03%, 18.59%��33.68%, �۱�ϩ���ƶ�3�ֿ������������ǿ��˳��Ϊөʯ>����ʯ>���ٿ�, ��ģ������������
3����
1.���ٿ�өʯ�ͷ���ʯ���ŵ�ģ�⺯����ΪLDA-CA-PZ����, �ض��ֱܷ�Ϊ277, 270��275 e V;�ܴ��ṹ����3�ֿ�������ھ�Ե�塣
2.���ٿ�өʯ�ͷ���ʯ3�ֿ����Caԭ�ӵ�̬�ܶ���ɺ�����, ���ٿ�ͷ���ʯ�����ܼ�������̬�ܶ���Ҫ������2p�������, өʯ�����ܼ������ļ۴���Ҫ��F2p������, ���, ���ٿ�ͷ���ʯ�ڲ��뻯ѧ��Ӧʱ��O�Ļ��Խ�ǿ, ��өʯ����F�Ļ��Խ�ǿ��
3.���ٿ�ͷ���ʯ���涼�мۼ�δ���͵ĸ�����, өʯ�����F-F����������, �������ӵ�ˮ�������ܱȱ�������ӵ�С, ��������ˮ��������Һ, ʹ�ñ���Ҳ�����ۼ�����������, �ڸ�ѡ�����б��ֳ����Ƶĸ�ѡ���ܡ�
4.��ˮ��ϵ��, �۱�ϩ���ƶ�3�ֿ��������ܾ���ֵ�Ĵ�С˳��Ϊ���ٿ�>����ʯ>өʯ, �ֱ�Ϊ-133.57, -178.54��-293.19 k J��mol-1, ˵���۱�ϩ������3�ֿ�����������ʹ�����ܵ���������, ��������ǿ��˳��Ϊөʯ>����ʯ>���ٿ�
5.�ڴ����︡ѡ������, ��p H=8.7~10ʱ, �۱�ϩ���ƶ�өʯ�ͷ���ʯ����������ǿ�ڰ��ٿ�, ����Ȼp H (δ����p H������) ������, ���ٿ�өʯ�ͷ���ʯ�Ļ����ʷֱ�Ϊ76.03%, 18.59%��33.68%, �۱�ϩ���ƶ�3�ֿ������������ǿ��˳��Ϊөʯ>����ʯ>���ٿ�, ��ģ�������������ɴ˿���˵���۱�ϩ�����п���ʵ�ְ��ٿ���өʯ������ʯ�ĸ�ѡ���롣
�����
[1] Qiu X Y, Dong T S.Modern Processing of Tungsten Ores[M].Beijing:Metallurgical Industry Press, 2012.111. (������, ������.�ִ��ٿ�ѡ��[M].����:ұ��ҵ������, 2012.111.)
[2] Liu Q G, Han Z Y, Guan Z G.Research progress on scheelite flotation technology[J].China Tungsten Industry, 2009, 24 (4) :23. (�����, ����Ԫ, �����.���ٿ�ѡ�о���չ[J].�й���ҵ, 2009, 24 (4) :23.)
[3] Xiao J H, Fan S P, Wang Z, Xu L H, Wang D Z.Comprehensive recovery of low grade tungsten-titanium polymetallic ore in Hubei[J].Chinese Journal of Rare Metals, 2013, 37 (4) :656. (Ф����, ��ɺƼ, ����, ������, ����־.������Ʒλ���Ѷ�������ۺϻ��������о�[J].ϡ�н���, 2013, 37 (4) :656.)
[4] Qiu T S, Chen X, Wen D X, Liao D H.Flotation process and flow tests of a refractory scheelite[J].Nonferrous Metals Science and Engineering, 2013, 4 (5) :48. (��͢ʡ, ����, �µ���, �ε»�.ij��ѡ���ٿ�ѡ���ռ����������о�[J].��ɫ������ѧ�빤��, 2013, 4 (5) :48.)
[5] An Z T, Luo X J.Tungsten dressing technology and its development[J].Mining Engineering, 2005, 5 (3) :29. (��ռ��, ��С��.��ѡ���ռ����չ[J].��ҵ����, 2005, 5 (3) :29.)
[6] Hu W B.Flotation[M].Beijing:Metallurgical Industry Press, 1983.9. (��Ϊ��.��ѡ[M].����:ұ��ҵ������, 1983.9.)
[7] Huo G S, Sun P M, Li H G, Li Y J, Zhao Z W, Sun Z M, Liu M S.A decomposing technique for scheelite concentrate and low-grade scheelite concentrate[J].Rare Metals, 2004, 23 (2) :115.
[8] Wang Y X, Cui G J.Tungsten (volume 1) [M].Ganzhou:Institute of Jiangxi Nonferrous Metals, 1979.7. (��ԥ��, ����.�� (�ϲ�) [M].����:������ɫұ���о���, 1979.7.)
[9] Li C F, Song X Y, Xue F K, Li Y, Zhang Y T, Li Z W.Study on beneficiation technology for low-grade wolframite and scheelite[J].Metal Mine, 2012 (5) :100. (����, ������, Ѧ����, ��Ө, ������, ��־ΰ.ij��Ʒλ�ڰ��ٿ�ѡ�������о�[J].������ɽ, 2012, (5) :100.)
[10] Marzari N, Vanderbilt D, Payne M C.Ensemble density-functional theory for ab-initio molecular dynamics of metals and finite-temperature insulators[J].Phy.Rev.Lett., 1997, 79 (7) :1337.
[11] Segall M D, Lindan P J, Probert M J.First principles simulation:ideas, illustrations and the CASTEP code[J].J.Phys.Cond.Matt., 2002, 14 (11) :2717.
[12] Chen Y, Chen J H, Guo J.Effect of natural impurities on the electronic structures and semiconducting properties of sphalerite[J].Acta Physico-Chimica Sinica, 2010, 26 (10) :2851. (����, �½���, ����.��Ȼ���ʶ���п����ӽṹ�Ͱ뵼�����ʵ�Ӱ��[J].������ѧѧ��, 2010, 26 (10) :2851.)
[13] Chen J H, Wang L, Chen Y.Density functional theory of effects of vacancy defects on electronic structure and flotation of galena[J].Chinese Journal of Nonferrous Metals, 2010, 20 (9) :1815. (�½���, ����, ����.��λȱ�ݶԷ�Ǧ����ӽṹ����ѡ��ΪӰ����ܶȷ�������[J].�й���ɫ����ѧ��, 2010, 20 (9) :1815.)
[14] Chen J H, Wang J M, Long X H, Guo J.First-principle theory on electronic structure of copper sulfides[J].Journal of Central South University (Science and Technology) , 2011, 42 (12) :3612. (�½���, ������, �����, ����.��ͭ������ӽṹ�ĵ�һ��ԭ���о�[J].���ϴ�ѧѧ�� (��Ȼ��ѧ��) , 2011, 42 (12) :3612.)
[15] Li Y Q, Chen J H, Chen Y, Guo J.Density functional theory calculation of surface properties of pyrite (100) with implications for flotation[J].Chinese Journal of Nonferrous Metals, 2011, 21 (4) :916. (������, �½���, ����, ����.������ (100) �������ʵ��ܶȷ������ۼ��㼰��Ը�ѡ��Ӱ��[J].�й���ɫ����ѧ��, 2011, 21 (4) :916.)
[16] Wen S M, Deng J S, Xian Y J.Theory analysis and vestigial information of surface relaxation of natural chalcopyrite mineral crystal[J].Transactions of Nonferrous Metals Society of China, 2013, 23 (3) :796.
[17] Cooper T G, Leeuw N H.A combined ab initio and atomistic simulation study of the surface and interfacial structures and energies of hydrated scheelite:introducing a Ca WO4potential model[J].Surface Science, 2003, (531) :159.
[18] Gao Z Y, Sun W, Hu Y H, Liu X W.Anisotropic surface broken bond properties and wettability of calcite and fluorite crystals[J].Transactions of Nonferrous Metals Society of China, 2012, 22 (5) :1203.
[19] Yuan P Q, Cheng Z M, Zhou Z M, Yuan W K.Zeta potential on the anti-scalant modified sub-micro calcite surface[J].Physicochemical and Engineering Aspects, 2008, 328 (1-3) :60.
[20] Minoru Itoh, Masami Fujita.Optical properties of scheelite and raspite Pb WO4crystals[J].Phys.Rev.B, 2000, 62 (19) :12825.
[21] Minoru Itoh, Dmitri L Alov, Masami Fujita.Exciton luminescence of scheelite and raspite structured Pb WO4crystals[J].Journal of Luminescence, 2000, (87-89) :1243.
[22] Anisimov V I, Aryasetiawan F, Lichtenstein A I.First-principles calculations of the electronic structure and spectra of strongly correlated systems:the LDA+U method[J].Journal of Physics:Condensed Matter, 1997, 9 (4) :767.
[23] Andrew J, John P Skinner, Lafemina.Structure and bonding of calcite:a theoretical study[J].American Mineralogist, 1994, 79:204.
[24] Gu B L, Wang X K.Solid State Physics[M].Beijing:Tsinghua University Press, 1989.110. (�˱���, ��ϲ��.��������ѧ[M].����:�廪��ѧ������, 1989.110.)
[25] Segal M D l, Shall R, Pickard C J.Population analysis of plane-wave electronic structure calculations of bulk materials[J].Phys.Rev.B, 1996, 54 (23) :16317.
[26] Zhao X X, Mi Y M.First-principle calculations on the atomic geometry and electronic states of CO monolayer on Cu (001) surface[J].Acta Physico-Chimica Sinica, 2008, 24 (1) :127. (������, ƃһ��.Cu (001) ����CO��������ṹ�͵���̬�ĵ�һ��ԭ���о�[J].������ѧѧ��, 2008, 24 (1) :127.)

