文章编号:1004-0609(2016)02-0415-08
萤石表面性质各向异性研究及进展
高跃升,高志勇,孙 伟
(中南大学 资源加工与生物工程学院,长沙 410083)
摘 要:基于萤石的晶体(表面)结构,研究并总结表面断裂键性质、解理及暴露面性质、表面(自由)能、润湿性、表面电性、吸附性等萤石表面物理化学性质的各向异性,讨论萤石晶体结构特征与表面性质之间的关系,分析不同表面性质之间的关联。结果表明:表面断裂键密度和表面能存在正相关性,表面断裂键密度可准确预测萤石的解理和暴露面性质,表面离子层结构差异、表面Ca质点及其断裂键差异可解释表面电性、润湿性、溶解性及吸附性的各向异性。
关键词:萤石;表面能;表面断裂键;各向异性;浮选
中图分类号:TD923 文献标志码:A
萤石(CaF2)是化学元素氟的最主要来源。氟的应用领域涵盖新能源、新材料、国防、光电、冶金、化工等产业,其作用不可替代。因此,国际上将萤石认定为战略保护矿物资源[1]。
自然界中,萤石通常与其他含钙镁矿物(如方解石、白钨矿、磷灰石、白云石等)紧密共生,浮选是从这类矿石中分离和回收萤石最主要的方法。浮选是利用不同矿物间表面性质差异进行分选的物理化学方法。但是,萤石与这些含钙镁脉石矿物皆为半可溶性盐类矿物,表面活性质点相同或相近,表面物理化学性质相似[2],造成萤石的高效浮选分离和回收存在较大难度。
萤石解理性质、表面能、表面电性、润湿性、表面吸附性等表面物理化学性质的各向异性研究可以更深刻地理解萤石与水溶液及浮选药剂的作用机制,对实现萤石与其他含钙镁矿物的高效浮选分离具有重要意义。萤石表面性质的各向异性鲜见系统性研究与总结,因此,本文作者研究并总结了近年来本课题组及国内外关于萤石表面性质各向异性的研究成果。
1 萤石结构
1.1 萤石晶体结构
萤石(CaF2)晶体属立方晶系,其空间群是Fm3m,晶胞常数为:a=b=c=0.5463 nm,α=β=γ=90°,Z=4。萤石的晶胞结构如图1所示。
Ca2+与周围8个F-离子结合成八配位,形成Ca-F8立方体。F-与周围4个Ca2+离子结合成4配位,形成正四面体。萤石晶体中只有一种Ca―F化学键类型,Ca―F键的键长为0.236 nm,Ca―F键的离子键分数为89.14%[3]。化学键的离子键分数大于50%,可认为该化学键为离子键,因此Ca―F键为强离子键。
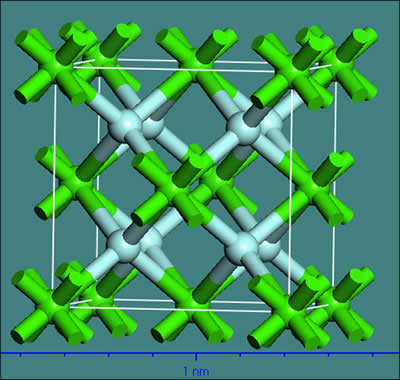
图1 萤石的晶胞结构(图中绿色为Ca,浅蓝色为F)
Fig. 1 Unit cell structure of fluorite (green is Ca, light blue is F)
1.2 萤石晶面结构
国内外研究较多的萤石晶面为{111}、{110}、{100}和{310}面,4个晶面的表面结构和原子的键合状态如图2所示。
2 萤石表面性质的各向异性
2.1 表面断裂键性质
矿物表面原子的断裂键性质属于矿物的本征性质。研究表明,矿物的表面断裂键性质可预测和验证矿物的解理性质和常见的暴露面,预测表面原子的化学反应活性[4]。
基于图2所示的萤石各晶面的表面结构和原子键合情况,GAO等[5]计算了萤石不同晶面的表面断裂键情况,结果如表1所列。由表1可知,萤石4个晶面表面断裂键密度(单位面积断裂键数)的大小排列依次如下:{100}、{310}、{110}、{111}。
2.2 解理及暴露面性质
矿物晶体在外力作用下严格沿着一定的结晶方向断裂,并且形成光滑平面的性质称为解理,这些光滑的平面称为解理面。在浮选前的破碎和细磨阶段,矿物的解理面一般平行于层间距较大的面及阴阳离子电性中和的面,并且沿矿物晶格内化学键强度最弱的方位产生[4]。
萤石{111}面方向,如图2(a)所示,F--Ca2+-F-电中性离子层(虚线框内离子层)依序分布,离子层内化学键和静电力很强,但是层间离子键的数目较少、强度很弱。由表1可知,{111}面表面断裂键密度是4个晶面中最小的,同时层间距最大,达到了0.3147 nm。因此,破碎和细磨时,沿{111}面最容易形成解理,{111}面是萤石最常见的解理面和暴露面。图3所示为萤石纯矿物粉末样的XRD谱,0.3153 nm处对应萤石{111}面,此处衍射峰的强度最大,说明{111}面的暴露程度最大,证明沿{111}面最容易产生解理。
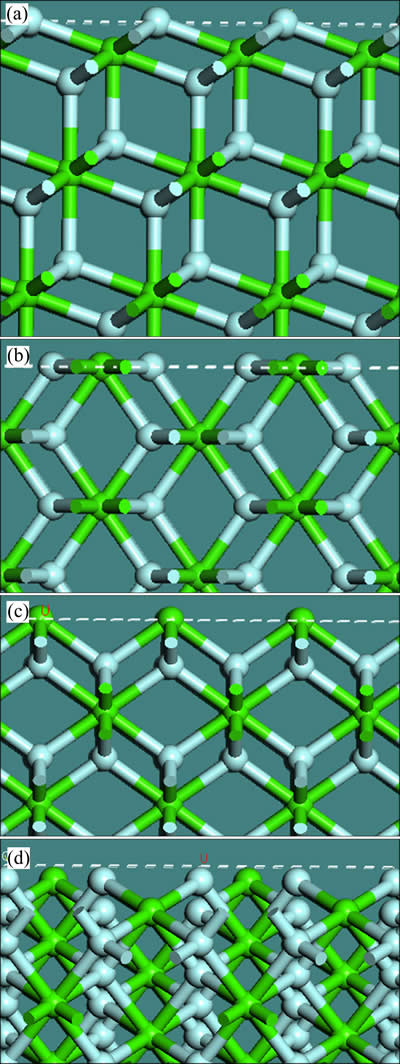
图2 萤石{111}、{110}、{100}、{310}面的表面结构及原子键合情况
Fig. 2 Structures and bonding states of {111}(a), {110}(b), {100}(c), {310}(d) surfaces of fluorite
表1 萤石各晶面表面断裂键计算[5]
Table 1 Calculation of surface broken bonds of fluorite[5]


图3 萤石纯矿物粉末样的XRD谱(d为层间距,nm)
Fig. 3 XRD pattern of powdered fluorite minerals for flotation test (d is interlayer spacing, nm)
萤石{110}面如图2(b)所示,表面离子层内Ca2+与F-以1:2的比例排布,{110}面为电性中和面。表1显示表面断裂键密度和层间距中等大小,因此{110}面可能暴露。在图3中,d=0.1929 nm处显示有较多的{110}面暴露,但是强度弱于{111}面的。
{100}面如图2(c)所示,Ca2+离子层与F-离子层依次排列,离子层之间化学键和静电作用较强,表面断裂键密度最大,层间距较小,因此该晶面因解理和断裂而暴露的概率非常小。图3中d=0.1366 nm处为 {100}面对应的微弱衍射峰,可见萤石纯矿物粉末样中该表面的暴露程度远小于{111}和{110}面的。
图2(d)表明,{310}面为电性中和面,单位面积断裂键数较{100}稍小,但是层间距最小。在图3中,d=0.1929 nm处为{310}面的衍射峰,表明{310}面的暴露程度略强于{100}面的,弱于{110}面的。
SARAVANAN等[6]基于萤石样品的XRD数据,使用最大熵法(MEM)计算表明,沿萤石[111]方向无电荷密度峰,而在[110]方向距离原点Ca原子1.93  处有一个0.48 e/3的电荷密度峰,在[100]方向距离原点Ca质点1.62 处有一个0.54 e/3的电荷密度峰。电荷密度峰值越大,表面Ca―F化学键的强度越大,该表面越不容易断裂。由此推断,{111}面最容易断裂,{110}面次之,{100}面较难断裂。与本课题组基于表面断裂键密度预测的萤石解理与暴露面吻合。
处有一个0.48 e/3的电荷密度峰,在[100]方向距离原点Ca质点1.62 处有一个0.54 e/3的电荷密度峰。电荷密度峰值越大,表面Ca―F化学键的强度越大,该表面越不容易断裂。由此推断,{111}面最容易断裂,{110}面次之,{100}面较难断裂。与本课题组基于表面断裂键密度预测的萤石解理与暴露面吻合。
2.3 表面能性质
表面能(等同于解理能),是指在外力作用下,矿物晶体沿某一晶面方向解理断裂成两个独立表面所需的能量,其大小取决于两个断裂表面间原子的相互作用强度。某个晶面的表面能越小,晶体沿该晶面方向越容易解理,解理形成的表面稳定性越高。
国外不同的研究小组计算了萤石常见晶面的表面能,计算结果如表2所列。由表2可知,{111}面的表面能最小,{110}面的次之,{100}和{310}面表面能最大。由此解释了{111}面最容易产生解理,是萤石的最常见暴露面,{110}次之,{100}和{310}面较难解理和断裂。
表2 文献报道的萤石各晶面表面能计算结果
Table 2 Literature reports on calculated surface energies of different fluorite surfaces

比较可知,基于表面能数据与基于表面断裂键密度预测的萤石解理性质相同,表明表面断裂键密度和表面能存在较强关联性。利用表1计算的萤石表面断裂键密度数据与文献报道的表面能数据作图,如图4所示。由图4可知,萤石表面能与表面断裂键密度呈正比例关系,随着表面断裂键密度的增加,表面能增加,决定系数R2大于0.9。表明表面单位面积断裂键数是影响甚至决定矿物表面能的关键因素。

图4 萤石表面断裂键密度与表面能之间的关系[5]
Fig. 4 Relationships between surface energy and surface broken bonds density of fluorite[5]
可见表面断裂键密度和表面能皆可以用来判断矿物表面的稳定性和解理性质。但是,相比复杂且耗时的表面能计算过程,表面断裂键密度的计算方法更简单、更省时,而且可以作为表面能计算值相对准确性的参考因素。
综上所述,对于萤石矿物,表面断裂键密度越小,表面化学键强度越小,表面能越小,沿该晶面方向越容易产生解理和断裂。萤石4个晶面的解理和暴露程度大小顺序如下:{111}、{110}、{310}、{100}。
GODINHO等[11]测定了萤石各晶面的溶解速率如表3所列。溶解速率差异首先归因于晶面的离子排布方式,同时存在Ca2+和F-离子的晶面({110}和{310}面如图2所示)溶解较快,而Ca2+和F-离子层依序排布的晶面({100}和{111}面如图2所示)溶解较慢。其次,离子排布方式相同的晶面,表面能越大,溶解速度越快。如{100}面的表面能较{111}面的大,{100}面溶解更快;{310}面的表面能较{110}面的大,{310}面溶解更快。此外,萤石晶体溶解行为主要是由于溶液的欠饱和状态造成的。
表3 萤石各晶面溶解速率[11]
Table 3 Dissolution rate of different fluorite surfaces[11]

2.4 表面润湿性
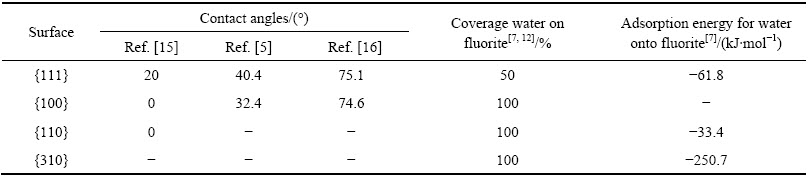
接触角大小是表征固体表面润湿性强度的一种方法。不同产地的萤石样品、不同的样品制备方法和测试手段,得到的接触角数值存在明显差异如表4所列。
由表4可知,萤石表现出一定的天然疏水性。JANCZUK等[12]研究表明,经过110 ℃干燥后测量的萤石接触角数值较50 ℃干燥后的接触角数值大,表明干燥温度越高,测得的萤石接触角越大。而且,萤石表面经Ca(OH)2溶液或HF溶液处理后的接触角较天然萤石的显著下降,这是由于萤石与极性溶液界面的酸碱分子间作用使萤石表面存在过剩的F-或Ca2+极性离子。由于F-比Ca2+具有更强的水合极性,因此萤石表面经HF溶液处理后的接触角较Ca(OH)2溶液处理后的更小,亲水性更强。ZAWALA等[13]研究表明,悬滴法比气泡捕捉法测得的萤石接触角大,越接近无色的萤石,疏水性越强。此外,接触角的测定值与测试水滴直径的大小以及浮选药剂浓度有关[14]。浮选药剂浓度不同,药剂分子在萤石表面的排列方式与吸附密度存在差异,一般低浓度下药剂分子在萤石表面形成单分子吸附层时,表面接触角最大,疏水性最强。
萤石表面润湿性存在各向异性,如表5所列。高志勇等[5]测算表明,萤石{111}和{100}面接触角分别为40.4°和32.4°,{111}面更疏水。ZHANG等[15]测得两个晶面的接触角分别为20°和0°。陆现彩等[16]也报道了类似的趋势。同一晶面的接触角测试值存在差异主要是由于样品的干燥温度不同造成的。接触角测试值较大的测试样品经50 ℃下干燥处理,而测试值较小的是在常温下干燥。
de LEEUW等[7]研究表明,水分子主要通过O原子与表面的Ca质点发生化学作用而在萤石表面吸附,H原子可与矿物表面F原子形成氢键。由图2可知,{111}面上最上层为F-层,Ca质点的暴露程度不高,且每个Ca质点只有一个断裂键,水分子吸附层在 {111}面只有50%的覆盖度[17]。{100}面最上层为Ca2+层,每个Ca质点具有4个断裂键,且Ca―Ca间距较大,水分子吸附层能够100%覆盖该表面。因此,{100}面较{111}面与水分子的作用更强,水分子在{100}面的覆盖度更大,{100}面更亲水。{310}面上有两种类型Ca质点,分别具有2个和4个未饱和键,Ca―Ca距离分别为0.544和0.86 nm,因此水分子吸附层可在{310}面形成100%的覆盖度。de LEEUW等[7]计算分析表明,单分子水吸附层与{310}面的作用能远大于{111}面的,如表5所列。
表4 文献中不同测试方法下萤石接触角测定结果
Table 4 Contact angle values of fluorite reported in Ref.[12-13]

表5 萤石各晶面的接触角及各晶面与水分子的作用能
Table 5 Contact angles of fluorite surfaces and interaction energies of water molecule with them
综上所述,萤石表面Ca质点的断裂键数(未饱和键数)越多,Ca质点的反应活性越强,与水分子的作用越强,表面亲水性越强。
2.5 表面电性
表面电性的影响因素可分为内因和外因。内因是指萤石表面离子层结构及各向异性等;外因是萤石所处的溶液化学环境和测试手段等(AFM、电泳)。
萤石矿物粉末样的零电点PZC在6.2~10.5之间[18],这是由于萤石样品所处的溶液条件不同造成的。BECRAFT等[19]研究表明,低pH溶液中,过剩的H+使萤石表面带正电;当pH增大至碱性时,OH-离子在萤石表面形成Ca(OH)2分子层并使萤石表面带负电。
萤石的表面电性存在各向异性。在pH=2~11范围内,萤石{100}面带正电(30mV左右)[15],{111}面带负电(-20 mV左右)[20],{110}面的等电点在pH为9.2左右[21],这与3个晶面的表面结构有关。在较短的测试时间内,萤石表面很难达到溶解和解离平衡,因此表面离子层的结构决定其表面电性。{111}面表层为F-层(见图2(a)),故带负电;{100}面表层为Ca2+层(见图2(c)),故荷正电;而{110}面则同时存在F-和Ca2+,与萤石粉末样品的表面电性类似,存在等电点。另外,矿物晶格畸变、点缺陷、晶格取代等也会引起表面电性的变化[22]。
2.6 表面吸附性
油酸(钠)和十二烷基磺酸钠(SDS)是萤石浮选较常用的两种捕收剂,两种捕收剂在萤石表面的吸附行为一直是矿物加工及相关领域的研究热点。
de LEEUW等[7]借助原子模拟,研究了与油酸具有同样极性羧酸基团的甲酸在萤石{110}、{111}、{310}面的吸附行为,发现吸附方式及吸附能存在各向异性,如表6所列。研究表明,脂肪酸类捕收剂的羧酸基团与矿物表面Ca质点可形成3种配位方式[23],分别为单配位(monodentate)、双配位(bidentate)和桥环配位(bridged),如图5所示。
表6 萤石各晶面Ca质点与甲酸的作用方式及作用能[8]
Table 6 Interaction model and energies of methanoic acid molecule with different fluorite surfaces[8]
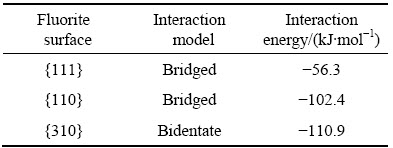

图5 油酸离子与含钙矿物表面Ca质点作用的3种构型示意图[23]
Fig. 5 Schematic diagrams of different binding conformations of oleate with surface calcium atoms[23]
研究表明,桥环配位构型最稳定,双配位次之,单配位较不稳定,且相邻两个Ca质点间距超过0.4 nm时,较难形成桥环配位。{310}面上相邻Ca质点间距为0.54和0.86 nm,甲酸分子的两个O只能与一个Ca质点形成双配位。{111}面上相邻Ca质点间距为0.385 nm,{110}面上的为0.385和0.544 nm,甲酸与这两个表面上相邻两个Ca形成桥环配位。在{111}面上,由于单位晶胞面积及相邻Ca质点距离较小,一个甲酸分子需要占据两个单位晶胞的面积范围,甲酸分子吸附层对该表面的覆盖度为50%,而对{310}面和{110}面的覆盖度为100%。
但是,用甲酸代替油酸,忽略了油酸长碳链的空间几何效应对羧酸基团与表面Ca质点的键合与配位方式的影响。MIELCZARSKI等[24]研究表明,油酸离子与萤石{111}面上的Ca质点形成双配位构型较稳定,与de LEEUW等[7]研究得到的甲酸-萤石吸附稳定构型存在差异。这可能是由于油酸的长疏水碳链对其羧酸基团与萤石表面Ca质点的作用存在一定限制作用,油酸不如甲酸分子那样可灵活地在矿物表面配位。
RAI等[25]利用分子模拟研究了油酸在萤石{111}面上的自组装单分子吸附层(SAMs)构型,发现油酸单分子层对萤石表面的覆盖度为67%,形成的单分子吸附层疏水性强。LU等[26]研究发现,在形成单分子吸附层之前,油酸离子在萤石表面的吸附密度大于在方解石表面的吸附密度,这与两种矿物常暴露面Ca2+质点密度差异有关。萤石常暴露面{111}和{100}面上Ca2+质点密度分别为12.9和11.1 μmol/m2,而在方解石{104}面上Ca密度仅为8.24 μmol/m2,表面Ca质点密度越大,油酸的吸附密度越大。因此,油酸作用下萤石的浮选效果优于方解石的。此外,萤石表面结构存在缺陷时有利于吸附层的形成[24]。
MIELCZARSKI等[27]发现,油酸单分子层在萤石表面吸收峰为1536 cm-1和1575 cm-1,分别对应双配位和单配位的吸附形式。当溶液呈酸性时,萤石表面吸附油酸分子层;当溶液呈碱性时,萤石表面吸附沉淀的油酸钙层,吸附速度更快,覆盖度更大。MIELCZARSKI等[28]还发现,油酸在萤石表面的吸附是一个动态过程,吸附行为受油酸浓度的影响。当浓度低于1×10-5 mol/L时,油酸分子在萤石表面形成不超过1个统计单分子吸附层;当浓度为1×10-5~ 1×10-4 mol/L时,动态性最突出,油酸分子先形成核心,接着往三维方向发展长大,形成厚度达到20个单分子层;当浓度超过1×10-4 mol/L时,油酸分子在溶液中形成胶束,吸附行为较弱。SREEDEVI等[29]计算不同浓度油酸吸附后萤石表面能的变化。随着油酸浓度增加,萤石表面活性Ca2+与油酸发生化学反应生成油酸钙沉淀,同时吸附造成表面Ca2+的减少,表面正电性减弱,萤石等电点随之降低。
 等[30]测算表明,天然萤石的表面能极性分量为8.09 mJ/m2,SDS吸附后萤石的表面能极性分量为2.00 mJ/m2。表明表面活性剂主要在萤石表面的活性质点上(主要是Ca质点)发生吸附。BECRAFT等[31]运用和频共振光谱(SFVS)观察研究表明,SDS浓度为3.5×10-4 mol/L时,萤石表面形成SDS单分子吸附层;当浓度为3.5×10-4~1×10-3 mol/L时,同时存在单分子和双分子吸附层;当浓度超过1×10-3 mol/L时,以双层吸附为主。
等[30]测算表明,天然萤石的表面能极性分量为8.09 mJ/m2,SDS吸附后萤石的表面能极性分量为2.00 mJ/m2。表明表面活性剂主要在萤石表面的活性质点上(主要是Ca质点)发生吸附。BECRAFT等[31]运用和频共振光谱(SFVS)观察研究表明,SDS浓度为3.5×10-4 mol/L时,萤石表面形成SDS单分子吸附层;当浓度为3.5×10-4~1×10-3 mol/L时,同时存在单分子和双分子吸附层;当浓度超过1×10-3 mol/L时,以双层吸附为主。
此外,张英等[32]基于密度泛函理论研究发现,水化体系中聚丙烯酸钠在萤石最常见暴露面{111}的吸附能为-293.29 kJ/mol,而在白钨矿{111}面和方解石常暴露面{104}面的吸附能分别是-133.57和-178.54 kJ/mol。可推测,聚丙烯酸钠对萤石的抑制效果最强,方解石的次之,白钨矿的最弱,与浮选试验结果一致。可见研究矿物的最常见暴露面性质,可更好地指导浮选药剂的选择和设计,更准确地解释浮选机理。
3 结论
1) 萤石各晶面的表面原子排布方式及表面断裂键性质存在各向异性。萤石表面断裂键密度与表面能呈正比例关系,且决定系数R2接近1,表明表面断裂键密度是影响表面能的关键因素。
2) 表面断裂键密度可用来预测矿物的解理性质和常见暴露面。萤石表面断裂键密度越小,表面能越小,沿该晶面越容易产生解离和断裂。萤石沿{111}面最容易解理,沿{110}面次之,{111}面是萤石最常见暴露面。
3) 表面断裂键密度与表面能影响表面润湿性、吸附性。表面断裂键密度越大,表面能越大,表面溶解性越强,表面亲水性越强。此外,制样方法、测试温度、测试方法对萤石润湿性的测试结果影响较大。
4) 萤石表面电性的各向异性与表面原子的排布方式有关。此外,溶液化学环境和测试手段(AFM、电泳)也会影响表面电性的测试结果。
5) 萤石表面的Ca活性质点密度、Ca质点的断裂键数、相邻Ca质点间距等差异影响浮选药剂在表面的吸附行为,矿物常见暴露面的性质可代表矿物的性质,可深入研究。此外,溶液pH、药剂浓度等也对吸附过程有较大影响。
REFERENCES
[1] 王文利, 白志民. 中国萤石资源及产业发展现状[J]. 金属矿山, 2014(3): 1-9.
WANG Wen-li, BAI Zhi-min. Fluorite resources in China and its industrial development status[J]. Metal Mine, 2014(3): 1-9.
[2] PRADIP, RAI B, RAO T K, SHAILAJA K, VETRIVEL R, MIELCZARSKI J, CASES J M. Molecular modeling of interactions of alkyl hydroxamates with calcium minerals[J]. Journal of Colloid and Interface Science, 2002, 256(1): 106-113.
[3] ZHUROVA E A, MAKSIMOY B A, SIMONOV V I, SOBOLEV B P. Structural studies of CaF2 (at 296 K) and Ca1-xPrxF2+x (at 296 and 170 K) crystals with x= 0.1: The changes in the fluorite anionic motif in the partial substitution of Ca2+ by Pr3+ cations[J]. Kristallogr, 1996, 41: 438-443.
[4] GAO Zhi-yong, SUN Wei, HU Yue-hua. Mineral cleavage nature and surface energy: Anisotropic surface broken bonds consideration[J]. Transactions of Nonferrous Metals Society of China, 2014, 24(9): 2930-2937.
[5] GAO Zhi-yong, SUN Wei, HU Yue-hua, LIU Xiao-wen. Anisotropic surface broken bond properties and wettability of calcite and fluorite crystals[J]. Transactions of Nonferrous Metals Society of China, 2012, 22(5): 1203-1208.
[6] SARAVANAN R, ISRAEL S. Bonding in fluorite compound CaF2 using MEM[J]. Physica B: Condensed Matter, 2004, 352(1): 220-226.
[7] de LEEUW N H, PARKER S C, RAO K H. Modeling the competitive adsorption of water and methanoic acid on calcite and fluorite surfaces[J]. Langmuir, 1998, 14(20): 5900-5906.
[8] de LEEUW N H, COOPER T G. A computational study of the surface structure and reactivity of calcium fluoride[J]. Journal of Materials Chemistry, 2003, 13(1): 93-101.
[9] JANICKI M J, DRZYMALA J, KOWALCZUK P B. Structure and surface energy of both fluorite halves after cleaving along selected crystallographic planes[J]. Physicochemical Problems of Mineral Processing, 2016, 52(1): 451-458.
[10] MALDONADO P, GODINHO J R A, EVINS L Z, OPPENEER P M. Ab initio prediction of surface stability of fluorite materials and experimental verification[J]. The Journal of Physical Chemistry C, 2013, 117(13): 6639-6650.
[11] GODINHO J R A, PIAZOLO S, EVINS L Z. Effect of surface orientation on dissolution rates and topography of CaF2[J]. Geochimica et Cosmochimica Acta, 2012, 86: 392-403.
[12] JANCZUK B, BRUQUE J M, GONZALEZ-MARTIN M L,DEL POZO J M. Wettability and surface tension of fluorite[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 1993, 75: 163-168.
[13] ZAWALA J, DRZYMALA J, MALYSA K. Natural hydrophobicity and flotation of fluorite[J]. Physicochemical Problems of Mineral Processing, 2007, 41: 5-11.
[14] DRELICH J, ATIA A A, YALAMANCHILI M R, MILLER J D. Formation and wetting characteristics of adsorbed layers of unsaturated carboxylic acids at a fluorite surface[J]. Journal of Colloid and Interface Science, 1996, 178(2): 720-732.
[15] ZHANG Xia, WANG Xu-ming, MILLER J D. Wetting of selected fluorite surfaces by water[J]. Surface Innovations, 2014, 3(1): 39-48.
[16] 陆现彩, 侯庆锋, 尹 琳, 赵连泽, 刘显东. 几种常见矿物的接触角测定及其讨论[J]. 岩石矿物学杂志, 2003, 22(4): 397-400.
LU Xian-cai, HOU Qing-feng, YIN Lin, ZHAO Lian-ze, LIU Xian-dong. Measurement of contact angles of several common minerals and its discussion[J]. Acta Petrologica et Mineralogica, 2003, 22(4): 397-400.
[17] FA Ke-qing, NGUYEN A V, MILLER J D. Interaction of calcium dioleate collector colloids with calcite and fluorite surfaces as revealed by AFM force measurements and molecular dynamics simulation[J]. International Journal of Mineral Processing, 2006, 81(3): 166-177.
[18] ZHANG Xia. Surface chemistry aspects of flourite and bastnaesite flotation systems[D]. Utah: The University of Utah, 2014.
[19] BECRAFT K A, RICHMOND G L. In situ vibrational spectroscopic studies of the CaF2/H2O interface[J]. Langmuir, 2001, 17(25): 7721-7724.
[20] MILLER J D, FA Ke-qing, CALARA J V, PARUCHURI V K. The surface charge of fluorite in the absence of surface carbonation[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2004, 238(1/3): 91-97.
[21] ASSEMI S, NALASKOWSKI J, MILLER J D, JOHNSON W P. Isoelectric point of fluorite by direct force measurements using atomic force microscopy[J]. Langmuir, 2006, 22(4): 1403-1405.
[22] KHENATA R, DAOUDI B, SAHNOUN M, BALTACHE M, RERAT A H, DRIZ M. Structural, electronic and optical properties of fluorite-type compounds[J]. The European Physical Journal B: Condensed Matter and Complex Systems, 2005, 47(1): 63-70.
[23] RAI B. Molecular modeling for the design of novel performance[M]. Florida: CRC Press, 2012: 140.
[24] MIELCZARSKI E, MIELCZARSKI J A, CASES J M, RAI B. Influence of solution conditions and mineral surface structure on the formation of oleate adsorption layers on fluorite[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2002, 205(1/2): 73-84.
[25] RAI B, PRADIP. Design of highly selective industrial performance chemicals: a molecular modelling approach[J]. Molecular Simulation, 2008, 34(10/15): 1209-1214.
[26] LU Yong-qiang, DRELICH J, MILLER J D. Oleate adsorption at an apatite surface studied by ex-situ FTIR internal reflection spectroscopy[J]. Journal of Colloid and Interface Science, 1998, 202(2): 462-476.
[27] MIELCZARSKI E, MIELCZARSKI J A, CASES J M. Molecular recognition effect in monolayer formation of oleate on fluorite[J]. Langmuir, 1998, 14(7): 1739-1747.
[28] MIELCZARSKI J A, MIELCZARSKI E, CASES J M. Dynamics of fluorite-oleate interactions[J]. Langmuir, 1999, 15(2): 500-508.
[29] SREEDEVI P, CHENNAKESAVULU K, KAVITHA M, PRABHAKAR S. Electrokinetic behavior of fluorite[J]. Journal of Dispersion Science and Technology, 2011, 32(9): 1306-1310.
[30] GONZALEZ-MARTIN M L, BRUQUE J M, GONZALEZ- CABALLERO F, PEREA-CARPIO R. The mechanism of adsorption of sodium dodecylsulfonate on fluorite and its surface free energy[J]. Applied Surface Science, 1996, 103(4): 395-402.
[31] BECRAFT K A, MOORE F G, RICHMOND G L. In-situ spectroscopic investigations of surfactant adsorption and water structure at the CaF2/aqueous solution interface[J]. Physical Chemistry Chemical Physics, 2004, 6(8): 1880-1889.
[32] 张 英, 王毓华, 胡岳华, 文书明, 王进明. 白钨矿与萤石、方解石电子结构的第一性原理研究[J]. 稀有金属, 2014, 38(6): 1106-1113.
ZHANG Ying, WANG Yu-hua, HU Yue-hua, WEN Shu-ming, WANG Jin-ming. First-principle theory calculation of electronic structures of scheelite, fluorite and calcite[J]. Chinese Journal of Rare Metals, 2014, 38(6): 1106-1113.
A review of anisotropic surface properties of fluorite
GAO Yue-sheng, GAO Zhi-yong, SUN Wei
(School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China)
Abstract: The research progress on anisotropic surface physicochemical properties of fluorite, including surface broken bonds, cleavage nature, surface (free) energy, surface charge, wettability and adsorption behavior, was reviewed in this work based on the crystal (surface) structure of fluorite. The relationships between crystal structure and surface properties were discussed, and the correlations among these surface properties were discussed. The results show that the positive relation between surface broken bonds density and surface energy of fluorite is found, and the former can be used to predict the cleavage nature and the commonly exposed surfaces. The surface Ca distribution feature and its broken (or unsaturated) bonds number can be used to explain the anisotropic surface charging properties, wettability, dissolution and adsorption behavior of fluorite.
Key words: fluorite; surface energy; surface broken bonds; anisotropy; flotation
Foundation item: Project(2015CX005) supported by Innovation Driven Plan of Central South University, China; Project (51404300) supported by National Natural Science Foundation of China; Project(2015M570689) supported by China Postdoctoral Science Foundation; Project(2015JJ3141) supported by Hunan Provincial Natural Science Foundation, China
Received date: 2015-07-09; Accepted date: 2015-12-09
Corresponding author: GAO Zhi-yong; Tel: +86-731-88830623; E-mail: zhiyong.gao@csu.edu.cn
(编辑 王 超)
基金项目:中南大学“创新驱动计划”项目(2015CX005);国家自然科学基金资助项目(51404300);中国博士后科学基金资助项目(2015M570689);湖南省自然科学基金资助项目(2015JJ3141)
收稿日期:2015-07-09;修订日期:2015-12-09
通信作者:高志勇,讲师,博士;电话:0731-88830623;E-mail: zhiyong.gao@csu.edu.cn

