网络首发时间: 2018-06-01 14:01
稀有金属 2019,43(05),551-555 DOI:10.13373/j.cnki.cjrm.xy18030020
Sm2 Fe17 的价电子结构及性能研究
宋春燕 胡利光 王书桓 赵定国
华北理工大学冶金与能源学院
华北理工大学唐山市特种冶金及材料制备重点实验室
摘 要:
依据固体与分子经验电子理论 (empirical electron theory, EET) , 利用键距差 (BLD) 法, 计算了Sm2 Fe17 晶体的价电子结构与理论键能、结合能。将由Sm2 Fe17 的晶体结构数据得到的19种实验键距值, Sm, Fe原子状态杂化参数输入根据键距差法编制的程序, 运行并筛选输出结果得到Sm2 Fe17 的价电子结构。Sm2 Fe17 的价电子分布表明:Sm2 Fe17 晶胞中Sm原子处于B种杂化第一阶, 晶格电子数为1; Fe原子处于C种杂化第九阶, 晶格电子数为0.0322;总的共价电子数为165.000。通过计算, Sm2 Fe17 晶体中第α =1的Fe-Fe键能最大, E 1 =262.6832 kJ・mol-1 , 是晶体熔化时需要破坏的主干键络, Sm2 Fe17 的理论结合能为39030.0175 kJ・mol-1 , 因此使Sm2 Fe17 相熔化需要较多的能量, 这些是SmFe合金宏观熔点比较高的主要原因;第α =19的Sm-Sm键能最小, E 19 =1.5563 kJ・mol-1 , 其晶体结构被破坏时需要的能量很小, 这是SmFe合金高温下易产生Sm原子氧化、挥发的主要原因;Sm2 Fe17 晶体中晶格电子总数较少且分布不均是SmFe合金产生脆性的主要原因。
关键词:
SmFe ;价电子结构 ;键能 ;结合能 ;
中图分类号: O73
作者简介: 宋春燕 (1978-) , 女, 河北石家庄人, 博士研究生, 研究方向:特种冶金及材料制备, E-mail:scy7825@163.com; *王书桓, 教授;电话:0315-8805009;E-mail:wshh88@ncst.edu.cn;
收稿日期: 2018-03-13
基金: 国家自然科学基金项目 (51574104); 唐山市特种冶金及材料制备基础创新团队项目 (17130202D) 资助;
Valence Electron Structure and Properties of Sm 2 Fe 17 Song Chunyan Hu Liguang Wang Shuhuan Zhao Dingguo
College of Metallurgy and Energy, North China University of Science and Technology
Tangshan Key Laboratory of Special Metallurgy and Material Preparation, North China University of Science and Technology
Abstract:
Based on empirical electron theory (EET) of solids and molecules, the valence electron structures, theoretical bond energies and binding energies of Sm2 Fe17 crystals were calculated using bond length difference (BLD) method. Sm and Fe atom state hybridization parameters, 19 experimental bond distance values obtained from the crystal structure data of Sm2 Fe17 were input into a program compiled according to the key distance difference method, and the output results were run and screened to obtain the valence electron structure of Sm2 Fe17 . The valence electron distribution of Sm2 Fe17 indicated that the Sm atom in Sm2 Fe17 unit cell was in the first order for B-type hybridization and the number of lattice electrons was 1; Fe atom was in the ninth order for C-type hybridization and the number of lattice electrons was 0.0322. Total number of covalent electrons was 165.000. It could be seen from the calculations, Fe-Fe bond energy of the first one in Sm2 Fe17 crystal was maximal, E 1 =262.6832 kJ・mol-1 , which was the backbone bond that needed to be destroyed in the melting of Sm2 Fe17 crystal. The theoretical binding energy of Sm2 Fe17 was 39030.0175 kJ・mol-1 . Therefore, it took more energy to melt the Sm2 Fe17 phase. These were the main reasons why the macro melting point of the SmFe alloy was relatively high. The Sm-Sm bond energy of the nineteenth bond was minimal, E 19 =1.5563 kJ・mol-1 . The energy required to destroy the crystal structure was small, which was the main reason for oxidation and volatilization of Sm atoms that were easily produced at high temperatures in SmFe alloy. The brittleness of SmFe alloy was mainly due to the lower summation of lattice electrons and the uneven distribution of the lattice electrons in Sm2 Fe17 crystal.
Keyword:
Sm2 Fe17 ; valence electron structure; bond energy; binding energy;
Received: 2018-03-13
目前, Sm2 Fe17 Nx 2 Fe14 B的磁体材料
[1 ,2 ,3 ]
, 具有优异的磁性能并可应用在高温领域
[4 ,5 ,6 ,7 ]
。 尽管Sm2 Fe17 Nx
[8 ]
。
国内外许多文章已经对钐铁合金的制备、 钐铁合金的渗氮处理进行了诸多报道, 这些大多都是基于热态实验进行的实验研究。 从原子以及价电子结构方面对Sm2 Fe17 研究钐铁合金各方面性质的报道还较少。 要想避免SmFe合金制备、 渗氮中的系列问题, 有必要对其晶胞以及价电子结构进行研究。 本文依据固体与分子经验电子理论 (empirical electron theory, EET) , 从价电子结构的角度利用EET理论中键能及结合能计算公式计算Sm2 Fe17 晶体的理论键能、 结合能, 研究SmFe合金的物理性质。
1 Sm2Fe17的晶体结构
2∶17型钐铁合金 (Sm2 Fe17 ) 的空间群为
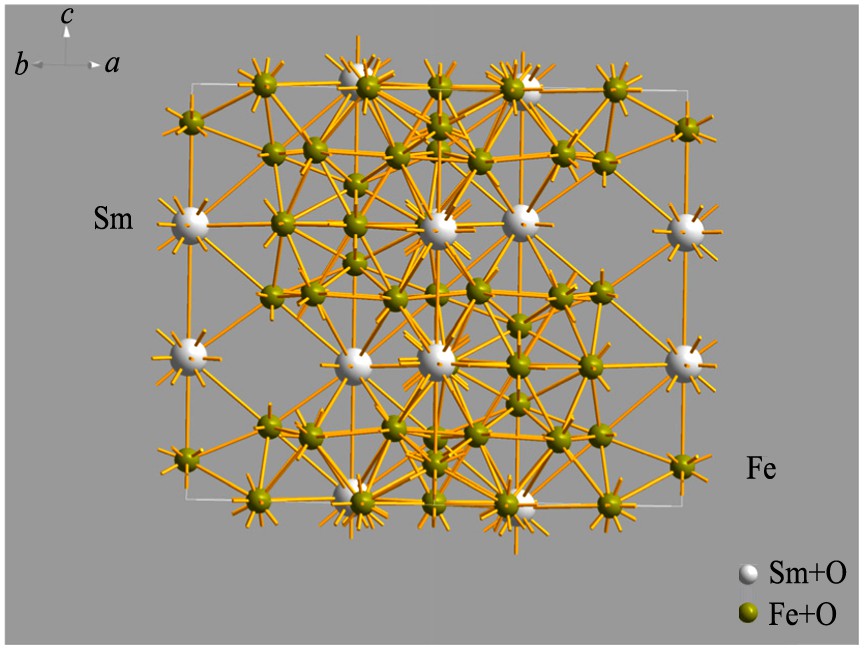
R ˉ 3 m ( 1 6 6 ) , 晶体结构属于三方晶系 (trigonal) , 点阵常数a =0.8558 (3) nm, c =1.2441 (3) nm; Z =3。 即单胞原子数为57 (共3个Sm2 Fe17 分子) , Sm2 Fe17 的结构式为Sm (6c) Fe (6c) Fe (9d) Fe (18f) Fe (18h) 。 各原子占位见表1, 其中Atom表示原子名称, Site表示各原子对称位置, x , y , z 表示相应原子的坐标信息, 晶胞结构图见图1。
图1 Sm2Fe17晶胞结构图
Fig.1 Cell structure diagram of Sm2 Fe17
表1 Sm2Fe17晶胞中原子占位坐标
Table 1 Space-occupying coordinates of atoms in Sm 2 Fe 17 cells
Atom
#
SITE
x y z SOF
H
1
6c
0
0
0.342 (2)
1
0
1
6c
0
0
0.094 (2)
1
0
2
9d
0.5
0
0.5
1
0
3
18f
0.291 (2)
0
0
1
0
4
18h
0.501 (2)
0.499 (2)
0.156 (2)
1
2 Sm2Fe17价电子结构及物性计算
2.1 Sm2Fe17的价电子结构
将Sm2 Fe17 晶胞参数输入Diamond可以得到晶胞结构图, 由Diamond软件
[9 ]
测量Sm2 Fe17 晶胞得到19种不可忽略的键 (<0.4 nm) 见表2, 其中Bonding number, Bonding atom, Bond ditance, Equivalent bond number分别表示成键序号、 成键原子、 实验键距与等同键数。
根据合金价电子结构的EET理论中的键距差 (BLD) 法
[10 ]
编制出计算程序, 将Sm, Fe原子杂化表
[11 ]
中的数据分别输入程序, 最后筛选出合理杂阶得到Sm2 Fe17 的价电子分布列于表3
[12 ]
。
Bond表示键名; I α 表示等同键数; D nα 表示实验键距;
ˉ D n α 表示理论键距; n α 表示共价电子数; |ΔD |表示键距差; ∑Ir, ∑n c , ∑n
D c 分别表示一个结构式里总的共价键数目、 总的共价电子数、 与扩散有关的物理量
[10 ]
; σ Sm , σ Fe 表示所选杂阶的序号, Sm有A/B两种杂化状态, Fe有A/B/C/D 4种杂化状态
[11 ]
。
表2 Sm2Fe17晶胞实验键距及等同键数
Table 2 Experimental bond distance and equivalent bond number of Sm 2 Fe 17 cells
Bonding
Bonding
Bond distanceD (n α ) /nm
Equivalent bond I α
Fe1-Fe1
0.23389
1
Fe2-Fe3
0.24389
6
Fe2-Fe4
0.24667
6
Fe3-Fe3
0.24904
2
Fe4-Fe4
0.24994
2
Fe3-Fe4
0.25509
4
Fe1-Fe2
0.26307
5
Fe3-Fe4
0.26393
4
Fe1-Fe4
0.26655
4
Fe1-Fe3
0.27513
8
Sm1-Fe3
0.30519
8
Sm1-Fe1
0.30854
2
Sm1-Fe4
0.30881
4
Sm1-Fe4
0.31980
4
Sm1-Fe4
0.32527
4
Sm1-Fe2
0.32957
5
Fe3-Fe3
0.35772
1
Fe4-Fe4
0.38817
1
Sm1-Sm1
0.39314
1
表3 Sm2Fe17的价电子分布
Table 3 Valence electron distribution of Sm 2 Fe 17
Bond
I α D nα /nmˉ D n α / n m
n α |ΔD |/nm
D n (Fe-Fe) 1
0.23389
0.23400
7.26796
0.00011
D n (Fe-Fe) 6
0.24389
0.24400
5.25494
0.00011
D n (Fe-Fe) 6
0.24667
0.24678
4.80189
0.00011
D n (Fe-Fe) 2
0.24904
0.24915
4.44664
0.00011
D n (Fe-Fe) 2
0.24994
0.25005
4.31874
0.00011
D n (Fe-Fe) 4
0.25509
0.25520
3.65444
0.00011
D n (Fe-Fe) 5
0.26307
0.26318
2.82115
0.00011
D n (Fe-Fe) 4
0.26393
0.26404
2.74356
0.00011
D n (Fe-Fe) 4
0.26655
0.26666
2.52007
0.00011
D n (Fe-Fe) 8
0.27513
0.27524
1.90795
0.00011
D n (Sm-Fe) 8
0.30519
0.30530
0.72280
0.00011
D n (Sm-Fe) 2
0.30854
0.30865
0.64839
0.00011
D n (Sm-Fe) 4
0.30881
0.30892
0.64274
0.00011
D n (Sm-Fe) 4
0.31980
0.31991
0.45003
0.00011
D n (Sm-Fe) 4
0.32527
0.32538
0.37688
0.00011
D n (Sm-Fe) 5
0.32957
0.32968
0.32782
0.00011
D n (Fe-Fe) 1
0.35772
0.35783
0.13102
0.00011
D n (Fe-Fe) 1
0.38817
0.38828
0.04880
0.00011
D n (Sm-Sm) 1
0.39314
0.39325
0.04189
0.00011
β =0.0710 ∑Ir=22.7024 ∑n c =165.0000 ∑n D C
σ Sm =B1 σ Fe =C9
2.2 Sm2Fe17的键能、 结合能计算
键能计算: 据文献[11]可知EET理论中两个相同原子形成的共价键键能公式为:
E α = b ? f ? n α D n α ? ? ? ? ? ? ? ? ? ( 1 )
式中: α 表示键名; n α 表示α 键上的共价电子对数; D n α f 表示共价电子成键能力; b 是一个键能计算参数, 按式
b = 7 5 n - 0 . 3 6 δ 求出;
f = √ α + √ 3 β + g √ 5 γ , α , β , γ 分别是成键的杂化轨道中s , p , d 成分, g对于4, 5, 6周期元素分别取1, 1.35, 1.70。
u , v 两个不同原子形成的共价键键能公式为:
E α = ˉ B α n α D ( n α ) ˉ F α ? ? ? ? ? ? ? ? ? ( 2 )
式中: nα (nα 的意义与 (1) 式相同;
ˉ B α = √ b u b v ;
ˉ F α = √ f u f v , 各参数值均取自文献
[
11 ]
。 经过计算, Sm2 Fe17 各键键能值列于表4中。
表4 Sm2Fe17的各键键能值
Table 4 Bond energy values of Sm 2 Fe 17
Bond
I α ˉ B α
ˉ F α
E α / (kJ・mol-1 )
D n (Fe-Fe) 1
262.6832
D n (Fe-Fe) 6
182.1434
D n (Fe-Fe) 6
164.5651
D n (Fe-Fe) 2
150.9408
D n (Fe-Fe) 2
146.0716
D n (Fe-Fe) 4
121.1088
D n (Fe-Fe) 5
90.6585
D n (Fe-Fe) 4
87.8780
D n (Fe-Fe) 4
79.9264
D n (Fe-Fe) 8
58.6261
D n (Sm-Fe) 8
41.4118
2.6843
26.3176
D n (Sm-Fe) 2
41.4118
2.6843
23.3520
D n (Sm-Fe) 4
41.4118
2.6843
23.1283
D n (Sm-Fe) 4
41.4118
2.6843
15.6375
D n (Sm-Fe) 4
41.4118
2.6843
12.8756
D n (Sm-Fe) 5
41.4118
2.6843
11.0534
D n (Fe-Fe) 1
3.0967
D n (Fe-Fe) 1
1.0629
D n (Sm-Sm) 1
1.5563
结合能计算: EET理论提出了普遍元素晶体结合能计算公式
[11 ]
:
ˉ E C = b [ ∑ α Ι α n α D ( n α ) f + n l ˉ D f ′ + a m 3 d - C W ] ? ? ? ? ? ? ? ? ? ( 3 )
徐万东等
[13 ]
在式 (3) 的基础上建立了过渡族金属化合物晶体的结合能计算公式:
ˉ E 0 C = ∑ α ˉ B α Ι α n α ˉ D ( n α ) ˉ F α + ˉ B l n l ˉ D ( n l ) f ′ + b u a m 3 d - b u C W ? ? ? ? ? ? ? ? ? ( 4 )
式中: b 是一个结合能能计算参数, 与键能有所不同;
ˉ B α = √ b u b v , u , v 是形成α键的两个原子;
ˉ B l = ( m + n ) √ b m u b n v , m , n 是分子式中u , v 的原子个数; n1 是晶格电子数; 等效键距
ˉ D = ∑ Ι α D ( n α ) ∑ Ι α ; 晶格成键能力
f ′ = √ 2 α ′ , α ′ = n l n Τ , n Τ 是总价电子数;
ˉ F α = 1 2 ( f u + f v ) , f u , f v 是形成α键的原子u, v的成键能力; b u , b v 是u, v元素晶体结合能的屏蔽系数值; 参数a =0.1542, 参数B 种杂化、 Fe原子C 种杂化无磁电子, 该项为0; 其他符号与键能公式中的一致。 经计算后Sm2 Fe17 的结合能为39030.0175 kJ・mol-1 。
3 分析与讨论
3.1价电子结构与Sm2Fe17稳定性的关系
Sm2 Fe17 的熔点较高, 但高温稳定性较差的原因可以从其价电子结构的特点方面分析。 EET理论提出
[8 ]
, 晶体熔化与晶体离解为单个原子不同, 熔化不需要破坏构成晶体的全部原子结构键, 只需将维持晶体中原子呈三维周期排列的主干键络破坏掉即可。 从表4中的键能计算结果可以看到, Sm2 Fe17 晶体的最大键能值为262.6832 kJ・mol-1 , 是维持晶体中原子呈三维周期排列的主干键。 因此使Sm2 Fe17 相熔化需要较多的能量, 宏观的熔点就比较高; 而最小键是两个Sm原子构成, 键能值为1.5563 kJ・mol-1 , 宏观表现为Sm2 Fe17 合金在常温、 非真空条件下易氧化, 高温条件下容易出现分解、 挥发的现象。
3.2价电子结构与Sm2Fe17脆性的关系
通过分析合金价电子结构可直接与其脆性相联系, 晶格电子主要影响金属与温度无关的韧性
[14 ]
。 Sm原子B 1 , Fe原子C 9 杂化状态的晶格电子n 1 总数为1.0322, 接近具有室温脆性的TiAl合金; 再者SmFe合金最近邻电子对数的不均匀分布直接导致室温应力作用下, 合金容易因弱键断裂而产生裂纹, 所以Sm2 Fe17 具有较高的脆性
[15 ,16 ]
。
4 结 论
1. Sm2 Fe17 晶胞中Sm原子处于B种杂化第一阶, 其晶格电子为1; Fe原子处于C 种杂化第九阶, 其晶格电子为0.0322。
2. Sm2 Fe17 晶体中第的Fe-Fe键能最大, E 1 =262.6832 kJ・mol-1 , 是晶体熔化时需要破坏的主干键络; 第α =19的Sm-Sm键能最小, E 1 =1.5563 kJ・mol-1 , 是SmFe合金高温下易产生Sm氧化、 挥发的主要原因。
3. Sm2 Fe17 晶体中晶格电子总数较少且分布不均是SmFe合金产生脆性的主要原因。
参考文献
[1] Qi M.Development of high performance rare earth magnets [J].Metallic Functional Materials, 2007, 14 (1) :41. (启明.高性能稀土磁体的开发 [J].金属功能材料, 2007, 14 (1) :41.)
[2] Li D Y, Duan H Q.Progress in industrialization of samarium iron nitrogen rare earth permanent magnet [J].China New Technologies and Products, 2012, (20) :146. (李大勇, 段焕强.钐铁氮稀土永磁材料产业化进展 [J].中国新技术新产品, 2012, (20) :146.)
[3] Yang Y C.Industrial development of new anisotropic rare earth permanent magnet materials [J].Advanced Materials Industry, 2011, (5) :21. (杨应昌.新型各向异性稀土永磁材料产业化开发进展 [J].新材料产业, 2011, (5) :21.)
[4] Hoffer G, Strnat K.Magnetocrystalline anisotropy of YCo5 and Y2 Co17 [J].IEEE Transactions on Magnetics, 2003, 2 (3) :487.
[5] Xue P, Guo X Y.The latest development of Sm2 Fe17 Nx 2 Fe17 Nx
[6] Coey J, Sun H.Improved magnetic properties by treatment of iron-based rare earth intermetallic compounds in anmonia [J].Journal of Magnetism & Magnetic Materials, 1990, 87 (3) :L251.
[7] Lu S, Yu D B, Li K S, Jin J L, Luo Y, Li H W.Effects of Fe content on structure of rapidly quenched and annealed Sm-Fe alloys [J].Chinese Journal of Rare Metals, 2013, 37 (5) :720. (卢硕, 于敦波, 李扩社, 靳金玲, 罗阳, 李红卫.Fe含量对快淬SmFe合金结构的影响 [J].稀有金属, 2013, 37 (5) :720.)
[8] Li J M, Cui C X, Han R P.Commercial status and development trends of Sm2 Fe17 Nx 2 Fe17 Nx
[9] Wu W X, Guo Y Q, Li A H, Li W.Analysis of valence electron structures and calculation of magnetic properties of Nd2 Fe14 B [J].Acta Physica Sinica, 2008, 57 (4) :2486. (吴文霞, 郭永权, 李安华, 李卫.Nd2 Fe14 B的价电子结构分析和磁性计算 [J].物理学报, 2008, 57 (4) :2486.)
[10] Liu Z L.Design of Electronic Structure and Composition of Alloy Composition [M].Jilin:Jilin Science and Technology Press Conference, 1990.245. (刘志林.合金价电子结构与成分设计 [M].吉林:吉林科学技术出版社, 1990.245.)
[11] Zhang R L.Empirical Electron Theory of Solids and Molecules [M].Jilin:Jilin Science and Technology Press Conference, 1993.275. (张瑞林.固体与分子经验电子理论 [M].吉林:吉林科学技术出版社, 1993.275.)
[12] Li L, Xing B.Valence electron structure analysis of diamond crystal growth from Fe-C system [J].Materials Chemistry & Physics, 2009, 117 (1) :276.
[13] Xu W D, Zhang R L, Yu R H.Calculation of binding energy of transition metal compounds crystals [J].Scientia Sinica (Series A) , 1988, (3) :101. (徐万东, 张瑞林, 余瑞璜.过渡金属化合物晶体结合能计算 [J].中国科学 (A辑) , 1988, (3) :101.)
[14] Wang Y D, Sun Z Q, Cai J, Chen G L.Valence electronic structure and brittleness of TiAl intermetallic compounds [J].Chinese Science Bulletin, 1991, (24) :1899. (王沿东, 孙祖庆, 蔡军, 陈国良.TiAl金属间化合物价电子结构及脆性 [J].科学通报, 1991, (24) :1899.)
[15] Song Q G, Zhao J P, Gu W F, Zhen D D, Guo Y R, Li Z P.Ductile and electronic properties of La-doped gamma-TiAl systems based on density functional theory [J].Acta Physica Sinica, 2017, 66 (6) :187. (宋庆功, 赵俊普, 顾威风, 甄丹丹, 郭艳蕊, 李泽朋.基于密度泛函理论的La掺杂γ-TiAl体系结构延性与电子性质 [J].物理学报, 2017, 66 (6) :187.)
[16] Cai J Y, Peng J Z, Yang X Z, Gray M F.A model of valence electron structure for embrittlement of TiAl [J].Materials Letters, 2008, 62 (24) :3957.