
A first-principles study on electronic structure and elastic properties of Al4Sr, Mg2Sr and Mg23Sr6 phases
ZHOU Dian-wu, LIU Jin-shui, PENG Ping
State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body,
Hunan University, Changsha 410082, China
Received 18 November 2010; accepted 29 March 2011
Abstract: The electronic structures and mechanical properties of Al4Sr, Mg2Sr and Mg23Sr6 phases were determined by the use of first-principles calculations. The calculated heat of formation and cohesive energy indicate that Al4Sr has the strongest alloying ability as well as the highest structural stability. The elastic parameters were calculated, and then the bulk modulus, shear modulus, elastic modulus and Poisson ratio were derived. The ductility and plasticity were discussed. The results show that Al4Sr and Mg2Sr phases both are ductile, on the contrary, Mg23Sr6 is brittle, and among the three phases, Mg2Sr is a phase with the best plasticity.
Key words: magnesium alloy; first-principles calculation; electronic structure; elastic property
1 Introduction
The interest in magnesium-based alloys is continuously increasing, especially because of their applications, e.g., automobile body materials, for weight reduction and higher fuel efficiency [1-2]. For the Mg-Al-based alloys, β-Mg17Al12 is an essential phase which plays an important role in strengthening crystal boundary and controlling high-temperature crystal running. However, the softening of the phase at the elevated temperature is detrimental to the creep property of the alloys. Therefore, the usage of these magnesium alloys in automobile industry is limited to non-critical parts. Calcium and strontium are two important additives used in magnesium alloys. However, Ca is detrimental since it results in the hot-cracking of the alloy [3]. On the other hand, Sr reduces the shrinkage and porosity of the alloy, and helps to abate the hot-cracking effect of Ca. Therefore, the Sr alloying magnesium alloys have drawn much attention in recent years.
Recent experiment investigations [4-6] have shown that strontium addition to the Mg-Al-based alloys improves greatly the heat resistance by forming Al4Sr,Mg2Sr and Mg23Sr6 phases. However, the alloying ability and the structural stability of these compounds containing strontium in Mg-Al-based alloys have not been well studied yet. The reason is that these compounds are often brittle and it is very difficult to prepare the sample for the measurements of the mechanical properties. As a result, the investigations of the mechanical properties have been performed only for a few cubic and simple hexagonal Laves phases by dynamic measurements [7]. Recent first-principles investigations of the elastic constants of metals based on the density functional theory give quite satisfactory results for the evaluation of bulk modulus, shear modulus and other elastic constants [8-9]. However, to the best of our knowledge, no systematic theoretical study has been performed on the electronic structure and mechanical properties of Al4Sr,Mg2Sr and Mg23Sr6 phases from first-principles.
In this work, the electronic structure and mechanical properties of Al4Sr, Mg2Sr and Mg23Sr6 phases are investigated by the use of first-principles method. The structures are optimized by full relaxations of the lattice parameters and atomic positions. The heat of formation and cohesive energy are calculated and discussed. The densities of states (DOS) are calculated to study the mechanism of structural stability. The elastic parameters Cij are calculated, the bulk modulus, shear modulus, elastic modulus and Poisson ratio are derived. The ductility, plasticity, and other mechanical properties of these compounds based on the calculated elastic properties are studied. The results give valuable estimation for the properties unavailable in experiments.
2 Method of computation
Cambridge serial total energy package (CASTEP) [10-11], a first-principles plane-wave pseudopotentials method based on density functional theory, was used in this work. CASTEP used a plane-wave basis set for the expansion of the single-particle Kohn-Sham wave- functions, and pseudopotentials to describe the computationally expensive electron-iron interaction, in which the exchange-correlation energy by the generalized gradient approximation (GGA) of Perdew was adopted for all elements in our models by adopting Perdew-Burke-Ernzerhof parameters [12-13]. Ultrasoft pseudopotentials [14-15] represented in reciprocal space was used. In the present calculations, the cutoff energy of wave functions (PWs), Ecut, was set at 330 eV. Sampling of the irreducible wedge of the Brillouin zone was performed with a 6×6×6 regular Monkhorst-Pack grid of special k-point. The finite basis set correction [16] and the Pulay scheme of density mixing [17] were applied for the evaluation of energy and stress. All lattice parameters and atomic positions in our model have been relaxed according to the total energy and force using the Broyden-Flecher-Goldfarb-Shanno [18] scheme, based on the cell optimization criterion (RMs force of 5.0×10-6 eV/?, stress of 0.01GPa, and displacement of 5.0×10-4 ?). The SCF tolerance is set as 5.0×10-7 eV. In our calculations of elastic constants of Al4Sr, Mg2Sr, Mg23Sr6 compounds, the generalized gradient approximation (GGA) of Perdew was used by adopting PW91.
3 Results and discussion
3.1 Structures and lattice constants
The structure parameters of Al4Sr,Mg2Sr and Mg23Sr6 phases are listed in Table 1, and these structures are shown in Fig. 1. The lattice constants of these structures are estimated from the minimized total energy, and the results are listed in Table 2. It can be seen that the obtained results of lattice constants are close to the available theoretical and experimental values [4, 19].The fairly good agreement between theoretical and experimental results show that the present calculations are highly reliable.
3.2 Heat of formation and cohesive energy
The heat of formation (?H) and the cohesive energy (Ecoh) of Al4Sr, Mg2Sr, Mg23Sr6 crystals are calculated by using the following expressions [21-22]:
 (1)
(1)
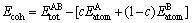 (2)
(2)
where 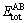 refers to the total energy/atom of the intermetallic compound;
refers to the total energy/atom of the intermetallic compound;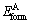 and
and 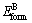 are the single atomic energies of pure constituents A and B in the elemental states; c refers to the atomic fraction of the constituent A;
are the single atomic energies of pure constituents A and B in the elemental states; c refers to the atomic fraction of the constituent A; 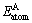 and
and 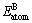 are the total energies of isolated atoms A and B. The obtained ?H calculated from Eq. (1) is also presented in Table 2.
are the total energies of isolated atoms A and B. The obtained ?H calculated from Eq. (1) is also presented in Table 2.
Table 1 Structure parameters of Al4Sr, Mg2Sr and Mg23Sr6 phases

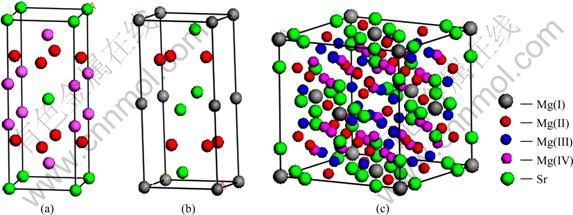
Fig. 1 Model of crystal cell of Al4Sr (a), Mg2Sr (b) and Mg23Sr6 (c)
Table 2 Lattice constants and formation heat of Al4Sr, Mg2Sr and Mg23Sr6 phases
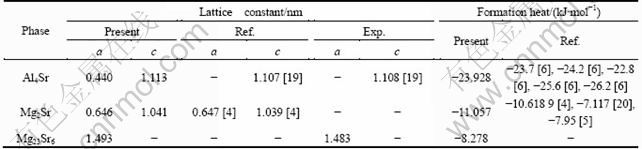
From Table 2 we can see that, the formation heat of Al4Sr is -23.928 kJ/mol, close to the corresponding calculation values of -23.7, -24.2, -22.8, -25.6 and -26.2 kJ/mol from Ref. [6]. The formation heat of Mg2Sr is -11.057 kJ/mol, in good agreement with the theoretical values of -10.618 9 from Ref. [4], -7.117 kJ/mol from Ref. [20] and -7.95 kJ/mol from Ref. [5]. The heats of formation of Al4Sr, Mg2Sr and Mg23Sr6 are all negative, which means that these phases are energetically stable. The negative heat of formation decreases from Mg23Sr6 to Mg2Sr to Al4Sr, indicating that the alloying ability increases from Al4Sr to Mg2Sr to Mg23Sr6 [23]. Al4Sr phase has the strongest alloying ability.
The stability of crystal is determined by its cohesive energy [24]. Generally, the cohesive energy can be defined as total energy released when isolated atoms combined into solid. Hence, the larger the absolute value is, the more stable the crystal structure is [24-25]. The obtained Ecoh calculated from Eq. (2) is shown Fig. 2. It is found that the present calculated results are -345.25 kJ/mol for Al4Sr, -156.80 kJ/mol for Mg2Sr and -152.91 kJ/mol for Mg23Sr6, respectively. The obtained results show that the Al4Sr phase is the most stable due to the highest Ecoh, and Mg2Sr is relatively stable, while the stability of Mg23Sr6 is the weakest due to the relatively lower Ecoh.
3.3 Electronic structures
Usually, structural stability of intermetallic compounds depends on the bonding electron orbital characteristics. For example, the strength of covalent bond is related to covalent electron orbital hybridization, while ionic bonds are decided by transfer charge for different atoms [26-27]. In the present work, further analysis of total and partial densities of states (DOS) (see Fig. 3) of Al4Sr,Mg2Sr and Mg23Sr6 phases are performed to reveal the structural stability mechanism of these compounds. The total and partial DOSs of Al4Sr,Mg2Sr and Mg23Sr6 crystal cells are plotted in Figs. 3(a), (b) and (c), respectively. Here, we can see that the main bonding peaks for these compounds basically locate in energy range from 0 to -10 eV, and originate from the contribution of valence electron numbers of Al(s), Al(p), Sr(s) and Sr(p) orbits for Al4Sr (see Fig. 3(a)), but for Mg2Sr and Mg23Sr6, those are the result of the bonding Mg(s), Mg(p), Sr(s) and Sr(p) (see Figs. 3(b) and (c)). Further analysis was done. It is found that for Al4Sr, covalent electron orbit hybridization takes place in 0- -2.5 eV energy range, which mainly is the weaker Sr(p)-Al(p) interaction, while for Mg2Sr, hybridization is thought as the sp states of Sr and the sp states of Mg, in addition, some resonance phenomena happen between Sr(s), Mg(s) and Mg(p). But for Mg23Sr6, hybridization range becomes broader compared with those of Al4Sr. Among three phases, there are most electronic states involved in hybridization and the strongest hybridization intensity for Mg23Sr6. Hence, from the perspective of covalent bond, the stability of Mg23Sr6 should be higher than that Mg2Sr or Al4Sr, which is not exactly the same order compared with that of the calculated cohesive energy. Hence, characteristics of ionic bonds need be considered for three phases.
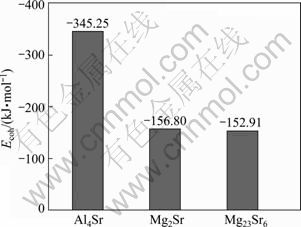
Fig. 2 Cohesive energy (Ecoh) of Al4Sr, Mg2Sr and Mg23Sr6 phases
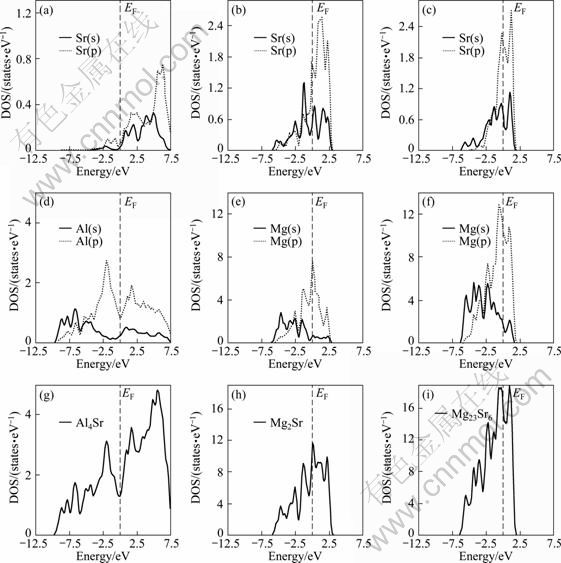
Fig. 3 Total and partial DOSs of Al4Sr (a, d, g), Mg2Sr (b, e, h) and Mg23Sr6 (c, f, i)
The calculated results of Mulliken electron occupation number for Al4Sr,Mg2Sr and Mg23Sr6 are listed in Table 3. It is found that the charge transfer phenomenon takes place from the Sr to Al atoms for Al4Sr, and the total number of the system is about 2.10, but for Mg2Sr and Mg23Sr6, the transfer charge from the Sr to Al atoms can be seen, the total number is about 3.88(0.97×4), 6.72(1.12×6), respectively. As far as Al4Sr, Mg2Sr and Mg23Sr6 are concerned, the system is not the same atomic number, which is 5, 12, 29 (see Table 1), respectively. If atom number of three phases is considered, the average transfer charge is approximately 0.420(2.10/5), 0.323(3.88/12), 0.232(6.72/29), indicating that the ionic bonds order from strong to weak is Al4Sr, Mg2Sr, Mg23Sr6, which is in good agreement with the cohesive energy calculations.
Table 3 Mulliken electronic populations of Al4Sr, Mg2Sr and Mg23Sr6

Calculations of the densities of states and Mulliken electronic populations for three phases show that the reason of Al4Sr with the highest structural stability attributes to Al4Sr phase having more the ionic bonds below Fermi level compared with those of Mg2Sr and Mg23Sr6 phases, while the structural stability of Mg2Sr is more than that of Mg23Sr6, which is the result of ionic and covalent bonds interaction.
3.4 Elastic properties
Elastic properties of Al4Sr,Mg2Sr and Mg23Sr6 phases, which are important for manufacturing Mg-Al- based alloys with strontium addition, are briefly discussed in this part. The calculated elastic constants for these intermetallics in the present work are listed in Table 4. As far as Mg23Sr6 phase, it is found that elastic constants satisfy the generalized elastic stability criteria for cubic crystals [7]: (C11+2C12)/3>0, C11+2C12>0 and C44>0. Hence, the computational elastic constants should be suitable. The bulk modulus B, shear modulus G and elastic modulus E are deduced according to the following formulae [28-29]:
 (3)
(3)
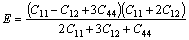 (4)
(4)
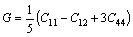 (5)
(5)
Poisson ratio ν is obtained from [7]
 (6)
(6)
Table 4 Calculated elastic constants of Al4Sr, Mg2Sr and Mg23Sr6
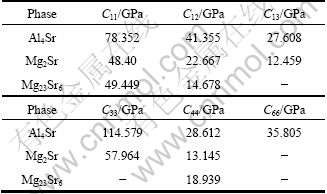
The obtained mechanical parameters of Al4Sr, Mg2Sr and Mg23Sr6 are listed in Table 5.
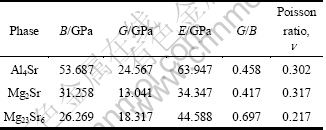
POUGH [30] introduced the ratio of the shear modulus to bulk modulus (G/B) of polycrystalline phases as prediction of the brittle and ductile behavior of materials. A high (low) G/B value is associated with ductility (brittleness). The critical value which separates ductility from brittleness is about 0.5. From G/B calculated in Table 5, it is found that Al4Sr and Mg2Sr both are ductile, on the contrary, Mg23Sr6 is brittle. The G/B value of Mg2Sr is the smallest, 0.417, indicating that Mg2Sr has very good ductility among the three phases.
Table 5 Moduli of there phases derived by this work from elastic constants
Besides G/B, it is found that C11-C12 and elastic modulus E are also very significant for the mechanical properties of materials [31]. The smaller the values of C11-C12 and elastic modulus E are, the better the plasticity is. Figure 4 illustrates the value of C11-C12 and elastic modulus E for Al4Sr, Mg2Sr and Mg23Sr6. From Fig. 4 we can see that Mg2Sr has lower values of elastic modulus E and C11-C12, implying better plasticity. On the contrary, Poisson ratio ν is used to quantify the stability of the crystal against shear, which usually ranges from -1 to 0.5. The larger the Poisson ratio is, the better the plasticity is. Most of the calculated Poisson ratios are very close to 0.25, which means that most of materials are with predominantly central interatomic forces [32]. Mg2Sr has larger Poisson ratio, showing that Mg2Sr is of good plasticity among the investigated compound. While for Mg23Sr6, the Poisson ratio is the smallest, corresponding to the poorest plasticity. All the results of the analysis on plasticity indicate that adding Sr to Mg-Al alloy can improve the ductility by forming Al4Sr and Mg2Sr phases.
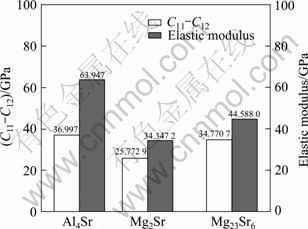
Fig. 4 Values of C11-C12 and elastic modulus E of Al4Sr, Mg2Sr and Mg23Sr6
4 Conclusions
1) The calculated heat of formation and cohesive energy show that Al4Sr has the strongest alloying ability and the highest structural stability.
2) Calculations of the densities of states (DOS) and Mulliken electronic populations show that the reason of Al4Sr with the highest structural stability attributes to Al4Sr phase having more the ionic bonds below Fermi level compared with those of Mg2Sr and Mg23Sr6 phases.
3) The calculated bulk modulus B, shear modulus G, elastic modulus E and Poisson ratio ν show that Al4Sr and Mg2Sr both are ductile, on the contrary, Mg23Sr6 is brittle, and among the three phases Mg2Sr is a phase with the best plasticity.
References
[1] MORDIKE B L, EBERT T. Magnesium properties-applications- potential [J]. Mater Sci Eng A, 2001, 302(1): 37-45.
[2] LUO A, PEKGULERYUZ M O. Cast magnesium alloys for elevated temperature applications [J]. J Mater Sci, 1994, 29(20): 5259-5271.
[3] HOLLRIGL-ROSTA F, JUST E, KOHLER J, MELZER H J. Magnesium in the Volkswagen [J]. Light Met Age, 1980, 38(7-8): 22-23, 26-28.
[4] ZHONG Y, SOFO J O, LUO A A, LIU Z K. Thermodynamics modeling of the Mg-Sr and Ca-Mg-Sr systems [J]. J Alloy Compd, 2006, 421: 172-178.
[5] ALJARRAH M, MEDRAJ M. Thermodynamic modelling of the Mg-Ca, Mg-Sr, Ca-Sr and Mg-Ca-Sr systems using the modified quasichemical model [J].Computer Coupling of Phase Diagrams and Thermochemistry, 2008, 32: 240-251.
[6] ZHONG Y, WOLVERTON C, CHANG Y A, LIU Z K. A combined CALPHAD/first-principles remodeling of the thermodynamics of Al-Sr: Unsuspected ground states energies by “rounding up the (un)usual suspects” [J]. Acta Mater, 2004, 52: 2739-2754.
[7] YU W Y, WANG N, XIAO X B, TANG B Y, PENG L M, DING W J. First-principles investigation of the binary AB2 type laves phase in Mg-Al-Ca alloy: Electronic structure and elastic properties [J]. Solid State Science, 2009, 11(8): 1400-1407.
[8] ANTON H, SCHMIDT P C. Theoretical investigations of the elastic constants in Laves phases [J]. Intermetallics, 1997, 5(6): 449-465.
[9] FAST L, WILLS J M, JOHANSSON B, ERIKSSON O. Elastic constants of hexagonal transition metals: Theory [J]. Phys Rev B, 1995, 51: 17431-17438.
[10] PAYNE M C, TETER M P, ALLAN D C, ARIAS T A, JOANNOPOULOS J D. Iterative minimization techniques for Ab initio total energy calculations: Molecular dynamics and cojugate gradients [J]. Rev Mod Phys, 1992, 64: 1045-1097.
[11] SEGALL M D, LINDAN P L D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14: 2717-2743.
[12] MARLO M, MILLMAN V. Density-functional study of bulk and surface properties of titanium nitride using different exchange- correlation functionals [J]. Phys Rev B, 2000, 62: 2899-2907.
[13] WHITE J A, BIRD D M. Implementation of gradient-corrected exchange-correlation potentials in Car-parrinello total-energy calculations [J]. Phys Rev B, 1994, 50: 4954-4957.
[14] VANDERBILT D. Soft self-consistent pseudopotentitals in a generalized eigenvalue formalism [J]. Phys Rev B, 1990, 41: 7892-7895.
[15] PERDEW J P, CHEVARY J A, VOSKO S H, JASKSON K A, PEDERSON M P, SINGH D J, FIOLHAIS C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation [J]. Phys Rev B, 1992, 46: 6671-6687.
[16] FRANSCIS G P, PAYNE M C. Finite basis set corrections to total energy pseudopotential calculations [J]. J Phys: Condens Matter, 1990, 2: 4395-4404.
[17] HAMMER B, HANSEN L B, NORKOV J K. Improved adsorption energetics within density-functional theory using revised Perdew- Burke-Ernzerh of functionals [J]. Phys Rev B, 1999, 59: 7413-7421.
[18] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Phys Rev B, 1976, 13: 5188-5192.
[19] ALJARRAH M, PARVEZ M A, LI J, ESSADIQI E, MEDRAJ M. Microstructural characterization of Mg-Al-Sr alloys [J]. Science and Technology of Advanced Materials, 2007, 8(4): 237-248.
[20] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV V, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl [J]. Acta Mater, 1998, 46(10): 3433-3442.
[21] SAHU B R. Electronic structure and bonding of ultralight LiMg [J]. Mater Sci Eng B, 1997, 49(1): 74-78.
[22] KING R C, KLEPPA O J. A thermochemical study of some selected Laves phases [J]. Acta Metall, 1964, 12(1): 87-97.
[23] SONG Y, GUO Z X, YANG R, LI D. First principles study of site substitution of ternary elements in NiAl [J]. Acta Mater, 2001, 49(9): 1647-1654.
[24] ZUBOV V I, TRETIAKOV N P. TEIXEIRA RABELO J N, SANCHEZ ORTIZ J F. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a Van der Waals crystal-fullerene C60 [J]. Phys Lett A, 1995, 198(5-6): 470-471.
[25] ZHANG X D. Analysis of relationships between cohesive energy, elastic moduli and lattice parameter of some high temperature intermetallics [J]. Intermetallics, 1995, 3: 137-140.
[26] FU C L, WANG X D, YE Y Y, HO K M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation [J]. Intermetallics, 1999, 7(2): 179-184.
[27] NYLEN J, GARCìA F J, MOSEL B D, P?TTGEN R, HǎUSSERMANN U. Structure relationships, phase stability and bonding of compounds PdSnn (n=2, 3, 4) [J]. Solid State Sci, 2004, 6(1): 147-155.
[28] HONG S Y, FU C L. Phase stability and elastic moduli of Cr2Nb by first-principles calculations [J]. Intermetallics, 1999, 7: 5-9.
[29] MEHL M J, OSBURN J E , PAPACONSTANTOPOULOS D A, KLEIN B M. Structural properties of ordered high-melting- temperature intermetallic alloys from first-principles total-energy calculations [J]. Phys Rev B, 1990, 41: 10311-10323.
[30] POUGH S F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals [J]. Philosophical Magazine, 1954, 45: 823-843.
[31] SUMER A, SMITH J F. Elastic constants of single-crystal CaMg2 [J]. J Appl Phys, 1962, 33: 2283-2286.
[32] MATTESINI M, AHUJA R, JOHANSSON B. Cubic Hf3N4 and Zr3N4: A class of hard materials [J]. Phys Rev B, 2003, 68: 184108-184112.
Al4Sr,Mg2Sr和Mg23Sr6相的电子结构与
弹性性能的第一原理研究
周惦武, 刘金水, 彭 平
湖南大学 汽车车身先进设计制造国家重点实验室,长沙 410082
摘 要:采用第一性原理计算Al4Sr,Mg2Sr和Mg23Sr6相的电子结构与弹性性能。合金形成热与结合能的计算结果显示Al4Sr具有最强的合金化形成能力和最高的结构稳定性。通过计算弹性常数、体模量、剪切模量、弹性模量和泊松比,讨论了体系的韧性与塑性行为。结果表明,Al4Sr和Mg2Sr为延性相,Mg23Sr6为脆性相,在3种金属间化合物中,Mg2Sr的塑性最好。
关键词:镁合金;第一性原理计算;电子结构;弹性性能
(Edited by LI Xiang-qun)
Foundation item: Project (200805321032) supported by Doctoral Fund of Ministry of Education of China; Project (51071065) supported by the National Natural Science Foundation of China; Project (71075003) supported by the Science Fund of State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, China
Corresponding author: ZHOU Dian-wu; Tel: +86-731-88711911; E-mail: ZDWe_mail@yahoo.com.cn
DOI: 10.1016/S1003-6326(11)61110-2

