中国有色金属学报 2004,(10),1752-1758 DOI:10.19476/j.ysxb.1004.0609.2004.10.023
TiO2 直接电解还原过程的研究
刘美凤 郭占成 卢维昌
中国科学院过程工程研究所,中国科学院过程工程研究所,中国科学院过程工程研究所 北京100080 ,北京100080 ,北京100080
摘 要:
采用SEM、EDS、XRD等方法对TiO2直接电化学还原产物进行分析,指出TiO2电极的还原是从外向内由高价到低价再到金属逐步进行的。对还原过程中电流、还原气相产物的分析结果表明:还原过程电流效率低,并且电流效率随电解时间延续而降低,阳极产物CO、CO2与阴极中间产物Ca发生副反应以及副反应产物炭黑造成电流短路是电流效率低的主要原因。提高电流效率的途径有两条:一是增大阴、阳极间距,减小副反应的发生;二是使电解池表面熔盐不断导出,或采取某一隔离措施,使副反应产生的炭黑在阴、阳极之间不造成电流短路。
关键词:
电化学还原 ;电流效率 ;TiO2 ;
中图分类号: TF823
作者简介: 刘美凤(1976),女,博士研究生.; 郭占成,研究员;电话:01062558489;E mail:guozc@home.ipe.ac.cn;
收稿日期: 2004-04-06
基金: 国家杰出青年科学基金资助项目(50225415);
Process of direct electrochemical reduction of TiO2
Abstract:
The products of electrochemical reduction of TiO2 were examined by SEM, EDS and XRD. The results show that TiO2 is reduced step by step from outside to inside and from high valence oxide to low valence oxide until to metal. The analysis of current and anode gases during electrolysis indicates that the average current efficiency is low and decreases with electrolysis time. The secondary reactions between CO, CO2 and Ca evolved at cathode and short circuit caused by the produced black carbon are the main reasons resulting in low current efficiency. There are two ways to improve the current efficiency. First, the distance between the anode and cathode should be increased (enough) in order to limit the secondary reaction. Second, the molten salt at the surface should be drained continuously or the anode and cathode should be separated to prevent short circuit caused by black carbon.
Keyword:
electrochemical reduction; current efficiency; TiO2 ;
Received: 2004-04-06
钛及其合金具有密度小、 比强度高、 耐腐蚀、 耐高温等优异性能。 世界钛工业正经历着由以航空航天为主要市场的单一模式, 向冶金、 能源、 交通、 化工、 生物医药等民用领域为重点发展的多元模式过渡。 钛将成为继铁、 铝之后崛起的“第三金属”, 21世纪将是钛的世纪
[1 ,2 ,3 ,4 ]
。
当前钛的生产采用金属热还原法, 主要为镁热还原法(Kroll法)
[3 ,4 ]
和钠热还原法( Hunter 法)
[4 ]
。 因为Hunter法比Kroll法生产成本高, 所以目前在工业中广泛应用的只有Kroll法。 Kroll 法从1948年开发当初就因其成本高、 还原效率低而受到批评。 半个世纪过去了, 尽管Kroll法经过了一系列的改进, 但是该工艺并没有发生根本的改变, 仍然是间歇操作, 小的改进并不能大幅度降低钛的价格。 因此只有开发新的、 低成本的连续化工艺才能从根本上解决钛生产成本高这一问题。 为此, 研究人员进行了大量的实验和研究
[5 ,6 ,7 ,8 ,9 ,10 ,11 ]
。
自从2000年我国旅英学者Chen GZ在著名期刊Nature发表题为Direct Electrochemical Reduction of Titanium Dioxide to Titanium in Molten Calcium Chloride一文后
[5 ]
, 国际上掀起了TiO2 电化学还原制备海绵钛的研究热潮。 TiO2 的直接电化学还原法是一种低能耗、 无污染的绿色生产新工艺。 目前关于TiO2 直接电解还原的反应机理还不明确, 并且对于电解还原过程的电流效率研究迄今未见有报道。 本文作者通过对TiO2 直接电解还原过程的研究, 发现电解还原过程中电流效率随时间而变化, 初步确定了该还原过程效率低的原因。
1 实验
1.1 实验设备及原料
实验装置如图1所示。 实验中采用电阻加热坩埚炉, 并配有可控硅温度控制器, 反应器为内置石墨坩埚(内径30~100 mm)的密封不锈钢筒; 电解电源为WYK-3010直流稳压电源。
图1 实验装置简图 Fig.1 Schematic reactor-electrolytic cell arrangements
实验中所用的电解原料为TiO2 , 工业级, 纯度大于99%, 平均粒度为0.372 μm; 熔盐为分析纯无水氯化钙, 含量大于96%, 其中除含水外, 其它杂质含量不超过0.5%。
电解过程在高纯氩气保护气氛下进行, 其中φ (Ar)>99.999%, φ (O2 )<3×10-6 , φ (H2 O)<3×10-6 。
主要分析设备为:X射线衍射仪D/max-RB (Japan, Rigaku, D/max-RB) 分析样品物相; JSM-6301F场发射扫描电镜分析样品断面形貌, 并配有X射线能谱仪(EDS)可进行元素分析; TC-136氮氧测定仪分析样品N、 O含量; JY38ICP光谱仪分析样品中其它元素含量; SP3400气相色谱分析气体组成。
1.2 实验步骤
TiO2 与胶粘剂按一定比例混合, 搅拌均匀后, 压制成直径为10 mm、 高5~10 mm的电极, 电极成型压力2~10 MPa。 在室温下放置48 h, 使其自然干燥, 然后在马弗炉中于900~1 200 ℃温度下烧结数小时后即可用于电解实验。 电解实验在如图1所示的装置中进行, 以高密度石墨坩埚壁作阳极, 二氧化钛电极作阴极, 在氩气(100 mL/min)保护下的氯化钙熔盐中进行电解。 电解过程分两个阶段进行, 首先直流低电压(小于Ca、 Ti析出电压)预电解, 目的是脱除熔盐中的水分和杂质, 然后提高电解电压至2.8~3.2 V进行析出Ti电解。 电解时间为4~20 h, 熔盐温度为900~1 000 ℃。 电解结束后, 电极在氩气保护下炉内自然冷却至室温。
1.3 样品检测
电解后的产物, 用水冲洗表面后, 在超声波辅助下用1%盐酸水溶液洗涤, 所得样品干燥后作断面SEM表面形态分析和EDS分析; 样品再经破碎、 研磨后用1%盐酸进一步洗涤, 干燥后样品进行氮、 氧含量和其它元素含量分析及XRD物相分析。 用盐酸溶液洗涤的目的是充分除去产物中的氯化钙, 以免影响样品检测结果。
2 结果与讨论
2.1还原产物断面形貌观察与组成变化
对于TiO2 电极未完全还原产物结构的研究有助于考察电解反应过程。 图2所示为某电解后产物
图2 电解产物的断面图 Fig.2 Cross section of partially reduced pellet
的断面宏观结构图。 其电解条件为:电解温度900 ℃, 电解电压3.1 V, 电解时间4 h。 图3所示为原始TiO2 电极及其未完全还原产物不同部位的SEM形貌。
从图2中可以看出, 整个电极分为3部分。 中心区域部分呈黑色, 结构比较致密, 如图3(b)所示, 但与原始电极断面形貌(图3(a))相比, 颗粒长大至2 μm左右, 孔隙率增大, 说明TiO2 已部分还原; XRD分析显示这一区域物相组成十分复杂, 主要以钛的低价氧化物和钛酸钙的形式存在。 EDS分析显示出这一区域有Ca、 Si、 Al、 Cl杂质元素存在, 可能主要是由高温熔盐渗入而未能洗涤分离的原因。
中间区域部分呈灰色, 颗粒进一步长大至8 μm左右, 孔隙率明显增大, 如图3(c)所示, 说明TiO2 已基本被还原成金属Ti; XRD分析显示这一区域主要是Ti; EDS分析显示出这一区域虽然仍有Ca、 Si、 Al、 Cl杂质元素存在 (其原因也是由高温熔盐渗入而未能完全洗涤分离) , 但含量很少, 主要构成物为金属Ti。 这一区域较中心区域杂质含量少, 是因为TiO2 还原导致产物孔隙大, 渗入的熔盐容易被洗涤除去。 原料TiO2 平均粒度仅为0.372 μm, 而该区域颗粒长大至8 μm左右, 其原因可能是TiO2 的还原反应是放热反应, 在反应的过程中发生Ti颗粒的烧结, 从而形成了图3(c)中的形貌。 随着氧的脱除以及钛颗粒的烧结, 孔隙率增加。
最外层肉眼观察已呈金属光泽, 说明TiO2 已彻底被还原成金属Ti, 颗粒呈现不规则形状相互连结在一起, 单个颗粒大小在10 μm左右, 如图3(d)所示。 XRD分析显示这一区域物相组成与中间区域相同, 主要以金属Ti的形式存在。 EDS分析已检测不出杂质元素。
对上述还原过程试样不同部位分别取样, 并洗涤彻底消除熔盐杂质影响, 分析得到其O、 N含量列于表1。 从不同部位测得的O含量来看, TiO2 电
表1 还原产物不同部位O、 N含量 Table 1 Concentration of oxygen and nitrogenin partially reduced product
Element
Inside
Middle
Outside
w (O)/%36.16
7.446
1.664
w (N)/%0.256
0.107
0.043
w (O)/w (Ti)1.695
0.241
0.050 6
图3 原始TiO2电极及其未完全还原产物不同部位的SEM形貌 Fig.3 SEM morphologies of starting TiO2 electrode and different parts of partially reduced product
极还原也是由外向内、 由高价向低价再到金属逐步进行的。 电极试样外层, TiO2 基本全部还原为金属Ti。 实际试样中的O含量可能比分析值低得多, 因为电解得到的金属Ti表面活性很高, 在制备分析样品过程中不可避免地会产生表面氧化; 最内层区的TiO2 还未完全还原到TiO, 中间层的TiO2 已部分还原为金属Ti。 原始电极试样N含量为0.042%, 电极试样还原过程中N向中心区域富集, 其原因有待于研究分析。
2.2 还原过程电流变化
关于TiO2 电解还原机理仍然是一个有争议的问题, 目前主要有两种解释, 一是剑桥大学的Fray等
[5 ,10 ,11 ,12 ]
提出的氧离子化机理, 他们认为TiO2 中的O离子进入熔盐迁移至阳极与C反应生成CO2 或CO; 一是日本的Okabe等
[7 ,8 ,13 ,14 ]
提出的钙热还原机理, 他们认为TiO2 的还原主要是由熔解在CaCl2 熔盐中的电化学还原产物Ca来完成, 两种机理总结如下:
1) 氧离子化
阴极反应为
TiOx x e- →
Ti(s)+x O2- (in CaCl2 ) (电化学反应) (1)
2) 钙热还原反应
Ca2+ +2e→Ca (电化学反应) (2)
Τ
i
Ο
2
+
2
C
a
?
→
2
C
a
?
2
+
+
2
Ο
?
2
-
+
Τ
i
(热还原反应) (3)
反应式中下划线表示该物质溶解在熔盐中。
阳极反应均为
O2- +C→CO+2e (电化学反应) (4)
2O2- +C→CO2 +4e (电化学反应) (5)
Fray等
[5 ]
之所以提出氧离子化机理, 主要是他们认为所用的电解电压2.8~3.2 V低于CaCl2 的分解电压3.213 V (900 ℃), 因而不会造成CaCl2 的分解, 所以不应该有Ca析出。 但是他们忽略了CaO的存在。 由于CaCl2 容易吸水, 在升温熔化时不可避免的会造成CaCl2 的水解(水解反应见2.3节), 熔盐中会有一定量CaO的存在。 根据热力学数据估计CaO分解为Ca和O2 的理论分解电压为2.71 V(900 ℃), 如果用炭阳极代替惰性阳极, 其分解电位可降低1.03 V。 因此, 在2.8~3.2 V电解时CaO会电解出Ca, 钙热还原反应可能会发生。 在实验过程中, 水洗电解试样时发现有气泡产生, 其气味与电石(CaC2 )水解气味一样。 XRD分析表明阴极附近有CaO存在, CaC2 和CaO的存在, 说明实验过程中有Ca析出, 因此验证了钙热还原机理的正确性。
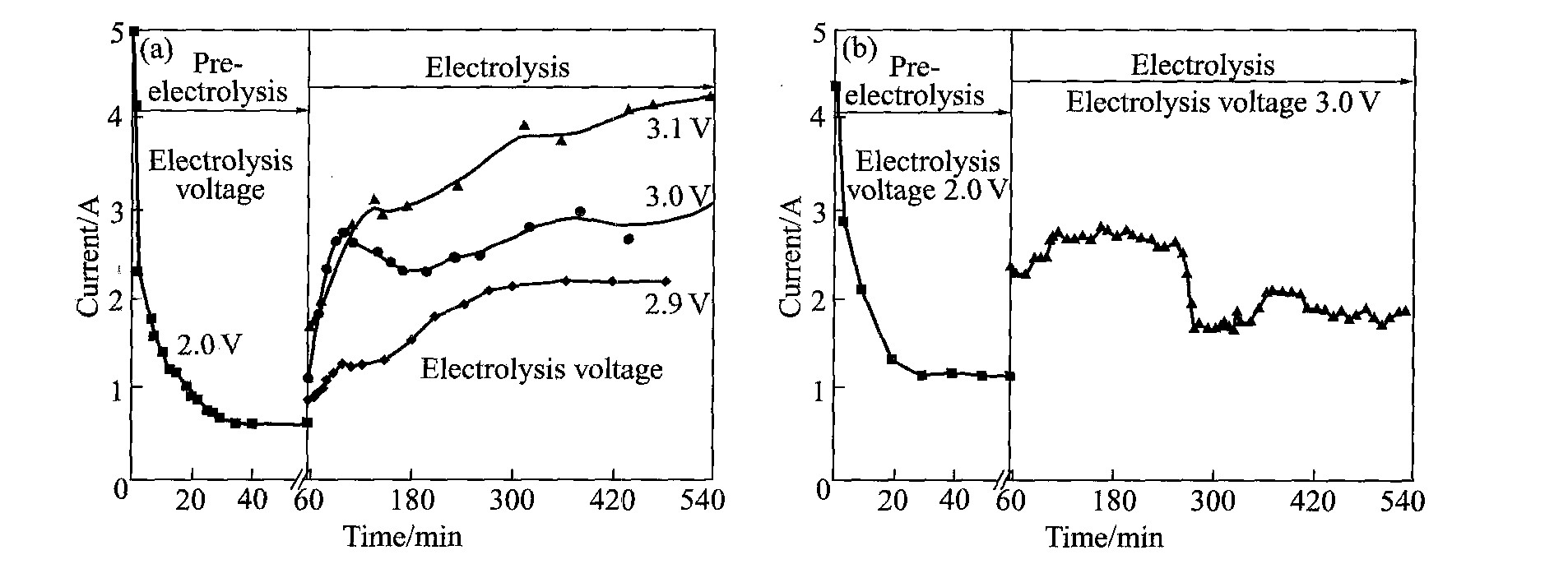
图4(a)所示是TiO2 电极在直径30 mm坩埚内CaCl2 熔盐中电解过程中的电流变化曲线。 在2 V恒电压下预电解过程中, 电流逐渐降低, 并很快减小到一恒定值。 预电解脱水后施以2.8~3.1 V某一恒定电压电解还原TiO2 , 电流随电解过程进行而增大; 电解电压越大, 电流也越大。 电解还原前期电流逐渐增加, 其原因可能是随电解进行电极还原反应面积增大所致。 如果TiO2 电解还原没有副反应, 随着电解反应的进行, 阴极中氧含量的不断降低, 电流应逐渐降低, 而实验结果却相反, 电流呈上升趋势, 电流效率很低, 说明副反应很严重。 电解实验结束后, 观察到熔盐表面有一层炭黑。 根据上述实验现象, 可以推测电解还原过程中发生的
图4 TiO2电极电解还原过程中的电流变化 Fig.4 Variation of current during electrolysis of TiO2 in different crucibles (a)—Crucible with d 30 mm; (b)—Crucible with d 100 mm
副反应。 如果坩埚很小, 即阴极和阳极间距很小, 阳极产生的CO或CO2 就容易与阴极产生的Ca发生反应:
CO+Ca→C+CaO (6)
CO2 +2Ca→C+2CaO (7)
2C+Ca→CaC2 (8)
炭黑的形成, 一是因为与氧离子作用生成气体造成C的脱落, 但最主要是由副反应(6)和(7)造成的。 由反应(6)和(7)生成的炭黑的存在, 一方面导致电流短路, 致使电解后期电流增加; 另一方面使副反应(8)发生, 导致电流效率降低。
为减小副反应的影响, 阴极和阳极间距应加大。 在直径100 mm的坩埚内重复上述实验, 即增大阴极和阳极间距, 电解过程电流变化如图4(b)所示, 电解后期电流降低至某一恒定值, 实验结束后熔盐表面没有明显的炭黑层, 水洗电极时也未发现气泡产生和C2 H2 气味。
2.3 电解还原气相产物组成
在100 mL/min 高纯氩气保护下采用SP3400气相色谱测定了电解过程中气相中CO、 CO2 、 O2 、 H2 的含量, 采用量程10 mg/m3 的氯气检气管检测电解过程中气相中的Cl2 含量。 电解过程中气相中CO、 CO2 、 O2 、 H2 浓度的变化如图5(a)所示, 但未检测到气相中有Cl2 存在。 电解过程没有Cl2 产生, 是由于电解电压3.0 V小于CaCl2 的分解电压3.213 V(900 ℃)。
预电解过程有H2 产生, 说明熔盐中有水分存在, 而且本实验在2 h的预电解脱水时间内, 熔盐中的水分并未完全脱除, 直到电解过程进行6 h后才基本将熔盐中的水分脱除。 如果预电解过程没有TiO2 的还原, 那么CO+2CO2 的浓度就应与H2 浓度相等, 而实际上CO+2CO2 的浓度远大于H2 浓度, 说明即使在预电解过程TiO2 也发生了电化学还原反应, 而且TiO2 还原速度远大于电解脱水速度。 在图5(a)中, 第一组数据, 即t =5 min时测得的CO、 CO2 浓度, 比第二组数据小, 但这并不说明预电解开始时反应速度小, 因为反应器的空腔约有500 mL, 电解反应产物反映到排出气中浓度大小有一个滞后时间, 结合图4(a)中预电解段电流的变化趋势看, 应当是开始时TiO2 电解还原和H2 O电解脱除反应最快。 预电解阶段虽然TiO2 被还原, 而且还原反应较快, 但在这一电解电压条件下, 从理论上讲, TiO2 不会被还原成金属钛, TiO2 只能被还原为钛的低价氧化物, 但这一推论还需要通过实验证实。 预电解过程尾气中不仅存在CO, 还产生了相当含量的CO2 , 但是在电解期, CO2 很少, 主要是CO, 并且CO产生量逐渐降低, 说明TiO2 电极有效还原反应速度随电解时间增加而降低。 而图4(a)所示同样条件下的电流并没有出现下降趋势, 甚至电流呈增加趋势, 这说明了电解副反应随时间增加而增大。
将图5(a)进行近似积分, 可估算出电解结束时产生的氧的总量约为0.047 8 mol, 并按H2 的产生量扣除H2 O电解产生的氧约0.0048 mol, 得到实际从阳极电解出氧的总量为0.043 0 mol(预电解段0.021 6 mol, 电解段0.021 4 mol)。 TiO2 电极试样重1.5 g, 其中氧为0.037 5 mol, 电解后分析其残
图5 TiO2电极电解还原过程中气体组成的变化(a)和电流效率的变化(b) Fig.5 Variation of current and concentration of CO, CO2, O2 and H2 during electrolysis of TiO2(a) and Current efficiency versus time(b) (TiO2 1.5 g, argon 100 mL/min, pre-electrolysis at 2.0 V and electrolysis at 3.0 V)
余氧含量为1.892%, 即从TiO2 电解还原出的氧为0.036 4 mol, 气体中排出的氧扣除H2 O电解出氧外仍大于TiO2 的失氧量, 说明除TiO2 和H2 O电解外, 有其它氧化物被电解还原。 熔盐中最有可能的氧化物是来自CaCl2 水解产生的CaO。 TiO2 电极在氯化钙熔盐的电解过程中, 尾气呈酸性, 但又无Cl2 存在, 说明熔盐内存在CaCl2 水解反应发生的可能性, 之所以不能完全肯定有水解反应发生, 是因为高温下CaCl2 熔盐有微量挥发, 挥发物CaCl2 遇水也呈酸性。 为进一步证实熔盐中存在CaCl2 的水解反应, 将实验用CaCl2 原料(1.5 V预电解10 h)和电解使用数次后的CaCl2 在300 ℃同时烘干脱水处理10 h, 分析其氧含量, 结果原料中氧含量为0.090 2%, 电解后氧含量为0.782 5%, 说明熔盐中确实存在氯化钙的水解反应:
H2 O+CaCl2 =CaO+2HCl (9)
Chen等
[15 ]
也指出在高温下存在氯化钙的水解反应, 为避免其干扰实验, 应对氯化钙的升温程序做严格规定。
本实验同时测得总通电量约32.5 Ah, 理论上应电解出氧约0.607 mol, 而实际上从TiO2 中电解出的氧约为0.036 4 mol , 电流效率仅为6.01%。 对图5(a)电解阶段电流大小和气体产物组成数据进行处理, 可以得到电解过程中电流效率的变化, 如图5(b)所示。 尽管总体上电流效率不高, 但电解过程前期电流效率还是较高的, 电流效率随电解过程的延续而降低。 在实际电解过程中, 电流随时间逐渐增加, 如果没有副反应发生, 电流效率应该是逐渐升高的, 这与实际得到的电流效率随时间下降相矛盾。 究其原因主要是化学副反应(6)和(7)的存在。 反应(6)和(7)的发生一方面使电化学反应(2)和化学反应(6)和(7)周而复始地循环进行, 另一方面造成熔盐表面覆盖炭层, 造成电流短路。 此外, 副反应(6)和(7)产生的炭黑, 还造成副反应(8)的发生。 由此看来, 要提高电流效率, 一方面阴、 阳极间距不能太小, 以减小副反应(6)和(7)的发生, 如图4(b)所示, 阴、 阳极间距增大后, 电流降低, 电流效率提高; 另一方面, 在工程上可使电解池表面熔盐不断导出, 或采取某一隔离措施, 使副反应(6)和(7)产生的炭黑在阴阳极之间形不成电流短路。
3 结论
1) CaCl2 熔盐中TiO2 电极的直接电解还原过程是由外到内从高价到低价再到金属逐渐进行的。 电解电流随电解电压增加而显著增大, 电化学还原反应速度随电解时间延续而快速降低。 电解还原过程的电流效率较低, 并且电流效率随电解时间延续而降低。
2) 阳极产物主要是CO、 CO2 , 并且阳极产物CO、 CO2 与阴极中间产物Ca发生副反应以及副反应产物炭黑造成电流短路是电解电流效率低的主要原因。 此外, 电解过程发生副反应生成CaC2 和熔盐中水分的电解也使电流效率降低。
3) 提高电流效率主要有两种方法:一是增大阴、 阳极间距, 以减小副反应的发生; 二是在工程上可使电解池表面熔盐不断导出, 或采取某一隔离措施, 使副反应产生的炭黑在阴阳极之间不形成电流短路。
参考文献
[1] 杨遇春.钛跨入新千年的金属巨人[J].中国工程科学,2002,4(3):2131.YANGYu chun.Titanium:Themetalgiantinthenewmillennium[J].EngineeringScience,2002,4(3):2131.
[2] 邓国珠.钛冶金的进展和发展方向探讨[J].稀有金属,2002,26(5):391396.DENGGuo Zhu.Discussionoftheachievementandthedevelopingtrendintitaniummetallurgy[J].ChineseJournalofRareMetals,2002,26(5):391396.
[3] 莫 畏,邓国珠,罗方承.钛冶金[M].北京:冶金工业出版社,1998.281334.MOWei,DENGGuo zhu,LUOFang cheng.TitaniumMetallurgy[M].Beijing:MetallurgicalIndustryPress,1998.281334.
[4] 孙 康.钛提取冶金物理化学[M].北京:冶金工业出版社,2001.49.SUNKang.TitaniumExtraction:MetallurgicalPhysicalChemistry[M].Beijing:MetallurgicalIndustryPress,2001.49.
[5] ChenGZ,FrayDJ,FarthingTW.Directelectrochemicalreductionoftitaniumdioxidetotitaniuminmoltencalciumchloride[J].Nature,2000,407:361364.
[6] OkabeTH,NikamiK,OnoK.Recenttopicsontitaniumrefiningprocess[J].BulletinoftheIronandSteelInstituteofJapan,2002,7(1):3945.
[7] SuzukiRO,TeranumaK,OnoK.Calciothermicreductionoftitaniumoxideandin situelectrolysisinmoltenCaCl2[J].MetallurgicalandMaterialsTransactionsB,2003,34B:287295.
[8] OkabeTH ,OdaR ,MitsudaY .Titaniumpowderbyperformreductionprocess(PRP) [J].JournalofAlloysandCom pounds, 2 0 0 4, 3 64:1 561 63 .
[9] 刘美凤 ,郭占成 .金属钛制备方法的新进展 [J].中国有色金属学报 , 2 0 0 3 , 1 3 (5) :1 2 3 81 2 45.LIUMei feng ,GUOZhan cheng .Newdevelopmentoftech nologiesforproducingtitanium [J].TheChineseJournalofNonferrousMetals, 2 0 0 3 , 1 3 (5) :1 2 3 81 2 45.
[1 0 ] FrayDJ.Emergingmoltensalttechnologiesformetalspro duction[J].JOM , 2 0 0 1 , 53 (1 0 ) :2 63 1 .
[1 1 ] FrayDJ,FarthingTW ,ChenGZ .RemovalofOxygenfromMetalOxidesandSolidSolutionsbyElectrolysisinAFusedSalt[P].InternationalPatentWO99/ 6463 8, 1 9991 2 1 6.
[1 2 ] ChenGZ ,FrayDJ,FarthingTW .Cathodicdeoxygenationofthealphacaseontitaniumandalloysinmoltencalciumchloride[J].MetallurgicalandMaterialsTransactionsB ,2 0 0 1 , 3 2B :1 0 41 1 0 52 .
[1 3 ] SuzukiRO ,InoueH .CalciothermicreductionoftitaniumoxideinmoltenCaCl2 [J].MetallurgicalandMaterialsTransactionsB , 2 0 0 3 , 3 4B :2 772 85.
[1 4] OnoK ,SuzukiRO .AnewconceptforproducingTisponge:calciothermicreduction[J].JOM , 2 0 0 2 , 54(2 ) :5961 .
[1 5] ChenGZ ,FrayDJ.Voltammetricstudiesoftheoxygen ti taniumbinarysysteminmoltencalciumchloride[J].JournaloftheElectrochemicalSociety , 2 0 0 2 , 1 49(1 1 ) :455467