ОДХВұаәЕЈә1004-0609(2008)09-1680-06
ЙБРҝҝуҪб№№AlNЎўAlPәНAlAsөДҪйөзәНөҜРФРФЦКөД
өЪТ»РФФӯАнСРҫҝ
Нх»АУС1, 2Ј¬Рм »Ы1Ј¬»ЖјТГф2Ј¬ХЕЕф»Ә3
(1. ЦРДПҙуС§ ОпАнҝЖС§УлјјКхС§ФәЈ¬іӨЙі 410083Ј»
2. ПжДПС§Фә ОпАнПөЈ¬і»ЦЭ 423000Ј»
3. ЦРДПҙуС§ ІДБПҝЖС§Ул№ӨіМС§ФәЈ¬іӨЙі 410083)
ХӘ ТӘЈәІЙУГөЪТ»РФФӯАнШНКЖЖҪГжІЁ·Ҫ·Ё¶ФЙБРҝҝуҪб№№AlNЎўAlPәНAlAsөДөзЧУҪб№№Ј¬ҪйөзәНөҜРФРФЦКҪшРРјЖЛгЈ¬Ҫб№ыұнГчЈә»щУЪГЬ¶ИәҜКэИЕ¶ҜАнВЫјЖЛгөДХвР©ІДБПөДҪйөзәНөҜРФРФЦКУлЖдЛьөЪТ»РФФӯАнәН·ЦЧУ¶ҜБҰС§јЖЛгЦөТ»ЦВРФҪПәГЈ¬ө«УлКөСйЦөЦ®јдУРТ»¶ЁөДІоТмЈ»ҫ§ёсІОКэөДУЕ»ҜЎўШНКЖөДСЎИЎәНҪ»»»№ШБӘПоөДСЎФс¶ј¶ФјЖЛгҪб№ыУРҪПҙуУ°ПмЎЈёщҫЭјЖЛгөДөҜРФіЈКэ»жЦЖБЛХвР©ІДБПМШХчЖҪГж(101)ГжөДөҜРФДЈБҝНјЈ¬ОӘХв·ҪГжІДБПөДБҰС§СРҫҝМṩІОҝјЎЈ
№ШјьҙКЈәөзЧУҪб№№Ј»ІЈ¶чУРР§өзәЙЈ»ҪйөзХЕБҝЈ»өҜРФіЈКэ
ЦРНј·ЦАаәЕЈәO 482Ј»O 472ЎЎЎЎ ОДПЧұкК¶ВлЈәA
First principle study on dielectric and elastic properties of
zinc-blended AlN, AlP and AlAs
WANG Huan-you1, 2, XU Hui1, HUANG Jia-min2, ZHANG Peng-hua3
(1. School of Physics Science and Technology, Central South University, Changsha 410083, China;
2. Department of Physics, Xiangnan University, Chenzhou 423000, China;
3. School of Material Science and Engineering, Central South University, Changsha 410083, China)
Abstract: With the pseudopotential plane-wave method of first principle, the electronic structure, dielectric and elastic properties of zinc-blende AlN, AlP and AlAs were studied. The results show that based on the density functional perturbation theory, the calculated results are in good agreement with other ab initio and molecular dynamics calculated values, but have definite difference with the experimental data. The calculated values are affected by lattice constant, selection of pseudopotential and selection of exchange-correlation energy. Based on the calculated elastic constant, the elastic moduli of characteristic plane (101) are figured, which offers reference to mechanical study of these materials afterward.
Key words: electronic structure; Born effective charge; dielectric tensor; elastic constant
ҪьДкАҙЈ¬ЙБРҝҝуҪб№№AlNЎўAlPәНAlAsТСөГөҪ№г·әСРҫҝ[1?4]Ј¬УЙУЪЛьГЗІ»ҪцКЗЦШТӘөДөзЧУәН№вөзЧУІДБПЈ¬¶шЗТКЗЦЖЧчТмЦКҪб№№Ўўі¬ҫ§ёсәНБҝЧУЪеөДЦШТӘіЙ ·Ц[5?6]ЎЈСРҫҝХвР©ҫ§МеөДөзЧУҪб№№ЎўҪйөзәНөҜРФРФЦКЈ¬ҝЙОӘ°лөјМеРВІДБПөДҝӘ·ўМṩАнВЫТАҫЭәНКөСйЦёөјЎЈУЙУЪәЬДСНЁ№э»ҜС§әНОпАнЖшПаіБ»эТФј°ёЯС№әПіЙҙуөДёЯЦКБҝөҘҫ§Ј¬ЛщТФ№ШУЪХвР©°лөјМеІДБПөДҪйөзәНөҜРФРФЦКөДКөСйКэҫЭұИҪПИұ·ҰЎЈОӘБЛ°пЦъАнҪвәНҝШЦЖХвР©ІДБПөДҪйөзәНөҜРФРФЦКЈ¬АнВЫјЖЛг·ЗіЈЦШТӘЎЈҪьДкАҙЈ¬¶ФХвР©°лөјМеІДБПөДҪйөзәН¶ҜБҰС§РФЦКТС№г·әөДІЙУГөЪТ»РФФӯАнҪшРРСРҫҝЈ¬ІўҪбәП¶аЦЦДЈРНәН·Ҫ·ЁҪшРРјЖЛгЈ¬АэИзјЫБҰДЈРНЎўҫшИИјьәЙДЈРНЎўёХРФАлЧУДЈРНТФј°¶іЙщЧУ·Ҫ·ЁЎўі¬Фӯ°ы·Ҫ·ЁЎўПЯРФПмУҰ·Ҫ·ЁөИЎЈУИЖдКЗФЪ1990ДкәуЈ¬ЛжЧЕГЬ¶ИәҜКэИЕ¶ҜАнВЫ(DFPT)[7?8]өДЦрІҪНкЙЖЈ¬МбёЯБЛ¶ФХвР©ІДБПРФЦКФӨІвөДЧјИ·РФЈ¬ЗТёГАајЖЛгІ»РиТӘЖдЛьКөСйЦөЎЈұҫОДЧчХЯ»щУЪГЬ¶ИәҜКэАнВЫКЧПИ¶ФЙБРҝҝуҪб№№AlNЎўAlPәНAlAsөДҫ§ёсІОКэҪшРРУЕ»ҜЈ¬И»әуТФУЕ»ҜәуөДҫ§ёсІОКэФЛУГDFPTјЖЛгХвР©ІДБПөДҪйөзәНөҜРФРФЦКЎЈ
1 ДЈРНәНјЖЛг·Ҫ·Ё
»щУЪГЬ¶И·әәҜАнВЫЈ¬ҙУөЪТ»ФӯАнШНКЖЖҪГжІЁ·Ҫ·Ёіц·ўЈ¬АыУГОДПЧ[9]ЦРөДЗшУтГЬ¶ИҪьЛЖ·Ҫ·ЁЈ¬№№ФмБЛөҘөзЧУКЖЦРҪ»»»№ШБӘПоЈ¬ІўАыУГTROULLIERәНMARTAIN(TM)·Ҫ·Ё[10]ІъЙъ·ЗҫЦУтЈ¬ДЈКШәгШНКЖЈ¬ТтОӘХвЦЦШНКЖДЬЙъіЙХэИ·өДөзәЙГЬ¶ИЈ¬ККәПЧчЧФЗўјЖЛгЎЈУҰУГГЬ¶ИәҜКэИЕ¶ҜАнВЫЈ¬КЧПИНкіЙҪб№№өДНкИ«ЛЙіЪЈ¬ҪУЧЕҪшРРПмУҰәҜКэјЖЛгЈ¬өГөҪХјУРМ¬ІЁәҜКэ№ШУЪФӯЧУО»ТЖЈ¬ҫщФИөзіЎәНУҰұдөДөЪТ»ҪЧОў·ЦЈ¬ЧоәуУГХвР©ЦөјЖЛг¶юҪЧОў·ЦПмУҰәҜКэХЕБҝЎЈјЖЛгЦРkҝХјд»э·ЦІЙУГMonhorst- Pack[11]·Ҫ°ёЈ¬Ҫ«ІјАпФЁЗш°ҙ8ЎБ8ЎБ8өД·ҪКҪ»®·ЦЎЈөзәЙГЬ¶ИІЙУГЛДГжМе»э·Ц·Ҫ·ЁјЖЛгЈ¬јЖЛгІҪіӨОӘ1.36ЎБ10?3 eVЎЈ
2 Ҫб№ыәНМЦВЫ
2.1 ҫ§ёсІОКэөДУЕ»ҜУлөзЧУ·ЦІј
AlЎўNЎўPәНAsөДөзЧУҪб№№·ЦұрОӘNe3s23p1Ј¬He2s22p3ЎўNe3s23p3әНAr4s24p3Ј¬ҙЛҙОІОУлјЖЛгөДAlјЫМ¬өзЧУОӘЧоНвІгөД3ёцөзЧУЈ¬БнНв3ёцФӘЛШөДјЫМ¬өзЧУОӘЧоНвІгөД5ёцөзЧУЎЈЙБРҝҝуҪб№№AlNЎўAlPәНAlAsөДҝХјдИәОӘF-43M(216)Ј¬ТхАлЧУәНСфАлЧУ·ЦұрТФГжРДБў·ҪҪб№№СШ¶ФҪЗПЯ1/4МЧ№№¶шіЙЎЈФЪұҫјЖЛгЦ®З°Ј¬КЧПИ¶ФЖдҫ§ёсіЈКэҪшРРУЕ»ҜЈ¬УЙУЪЦ»УРТ»ёцҫ§ёсіЈКэЈ¬ҪцРиАыУГДЬБҝЧоөНФӯАн¶ФMonkhorst-PackёсЧУәНҪШ¶ПДЬЧчКХБІІвКФЎЈјЖЛгөГөҪЈ¬Ждҫ§ёсіЈКэ·ЦұрОӘ4.337Ўў5.448әН5.607 ?Ј¬Ҫ«ЖдУлКөСйЦө4.38[12]Ўў5.465[13]әН5.655 ? [14]ұИҪПЈ¬ҙЛҙОөДУЕ»ҜЦөУлЖдЛьКөСйЦө·ЗіЈҪУҪьЈ¬ОуІоҪцФЪ1%ТФДЪЎЈ
»щУЪјЖЛгөДАнВЫёсЧУЦөЈ¬јЖЛгІў·Цұр»жіц3ЦЦ»ҜәПОп°лөјМеСШ[111]·ҪПтөзәЙГЬ¶И·ЦІјЈ¬ИзНј1ЛщКҫЎЈФЪНј1ЦРҪ«ҫ§°ы¶ФҪЗПЯЧчБЛ№йТ»»ҜҙҰАнЎЈҙУНј1ҝЙТФҝҙіцЈ¬ЛжЧЕТхАлЧУФӯЧУРтКэөДФцјУЈ¬Фӯөг(СфАлЧУ)өҪ¶ФҪЗПЯ1/4ТхАлЧУЦРјдөДөзәЙГЬ¶ИјхРЎЈ¬ХвЛөГчЛжЧЕТхАлЧУФӯЧУРтКэөДФцјУЈ¬УЙУЪҫ§ёсіЈКэөДФцјУәНФӯЧУөзёәРФөДјхРЎЈ¬К№өГАлЧУіЙ·ЦјхРЎЈ¬Ал»Ҝ¶ИҪөөНЈ¬ҪбәПБҰПВҪөЎЈН¬КұЈ¬ФЪНјЦРҝЙТФҝҙіцУЙУЪФЪШНКЖјЖЛгЦРҪцҝјВЗБЛјЫМ¬өзЧУЈ¬Г»УР°ьАЁәЛөзәЙЈ¬ЛщТФФЪФӯЧУәЛІҝО»јёәхГ»УРөзәЙ·ЦІјЎЈ
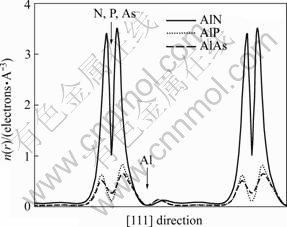
Нј1 ЙБРҝҝуҪб№№AlN, AlPәНAlAsСШ[111]·ҪПтөДјЫөзәЙГЬ¶И·ЦІј
Fig.1 Line plots of valence-charge density of zinc-blende AlN, AlP and AlAs along [111] direction
»щУЪјЖЛгөДҫ§ёсіЈКэЈ¬јЖЛг3ЦЦ»ҜәПОп°лөјМеөДөзЧУМ¬ГЬ¶ИЈ¬Ҫб№ыИзНј2ЛщКҫЎЈҙУНј2ҝЙҝҙіцЈ¬ЛжЧЕТхАлЧУФӯЧУРтКэөДФцјУЈ¬Жд»ҜәПОпөД·СГЬДЬПтөНДЬ·ҪПтТЖ¶ҜЈ¬өҘөзЧУМ¬ЧоёЯХјҫЭ№мөАәНЧоөНОҙХјҫЭ№мөАөДДЬј¶Іо¶ФAlNЎўAlPәНAlAs·ЦұрОӘ3.37Ўў1.61әН1.25 eVЈ¬ұИКөСйІв¶ЁөДҪыҙшҝн¶ИЦө (AlPәНAlAs·ЦұрОӘ2.50әН2.32 eV[15]Ј¬ҫЭұҫОДЧчХЯЛщЦӘЈ¬»№Г»УРAlNөДКөСйҙшП¶ЦөұЁөА)ТӘРЎЈ¬ХвКЗУЙУЪУГLDA·Ҫ·ЁЗуҪвјӨ·ўМ¬ДЬБҝКұұҫЙнҙжФЪөДІ»ЧгЈ¬өјЦВАнВЫјЖЛгөДҪыҙшҝн¶ИөНУЪКөСйЦө[16]ЎЈ
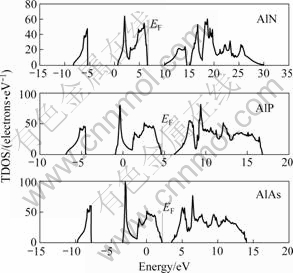
Нј2 ЙБРҝҝуҪб№№AlN, AlPәНAlAsөДЧЬөзЧУМ¬ГЬ¶И
Fig.2 Total density of states (TDOS) of zinc-blende AlN, AlP and AlAs
2.2 »ҜәПОпөДҪйөзРФЦК
ҪйөзіЈКэЦчТӘКЬБҪёцТтКэУ°ПмЈ¬Т»ёцКЗөзЧУЈ¬БнТ»ёцКЗЙщЧУЎЈөзЧУ¶ФҪйөзіЈКэөДУ°ПмВъЧгТФПВ№ШПө КҪ[17]

өзЧУ¶ФҪйөзіЈКэөД№ұПЧЦ»УРФЪ ІЕұдөГЦШТӘЈ¬ІўҙжФЪПВБР№ШПөКҪ[17]Јә
ІЕұдөГЦШТӘЈ¬ІўҙжФЪПВБР№ШПөКҪ[17]Јә

ІЈ¶чУРР§өзәЙКЗФЪНвіЎОӘБгөДМхјюПВЈ¬УЙУЪФӯЧУөДО»ТЖТэЖрөДәк№Ыј«»ҜөДұд»ҜУлФӯЧУО»ТЖөДұИВКЈ¬ҝЙұнКҫОӘ

КөјКЙПЈ¬ІЈ¶чУРР§өзәЙКЗ№вС§ЙщЧУУлөзіЎсоәПөДҪб№ыЎЈН¬СщЈ¬ІЈ¶чУРР§өзәЙҝЙ·ЦҪвОӘАлЧУәНөзЧУөД№ұПЧЈ¬јҙ

¶ФAlNЎўAlPәНAlAsҪйөзРФЦКөДјЖЛг№І·ЦИзПВ3ІҪНкіЙЎЈ
Step 1 BerryПа·Ҫ·ЁЧчТ»УРПЮөзіЎПВөД»щМ¬јЖЛгЈ¬ХвТ»ІҪУЙABINT[18]Инјю°ьЦҙРРЈ¬өГөҪЦШТӘөДј«»ҜіЎІјВеәХәҜКэЎЈ
Step 2 ҪшРРПЯРФПмУҰјЖЛгөГөҪІјВеәХәҜКэөДөЪТ»ҪЧПмУҰЎЈ
Step 3 АыУГөЪТ»ҪЧІјВеәХәҜКэПЯРОПмУҰјЖЛгҪйөзәНІЈ¶чУРР§өзәЙХЕБҝЎЈ
ФЪ¶Ф3ЦЦ»ҜәПОп°лөјМеөДҪйөзРФЦКјЖЛгЦРЈ¬І»Ҫц¶ФІЁәҜКэөДЖҪ·ҪУаКэЙи¶ЁТ»ёцЧоҙуФКРнЦө1ЎБ10?20Ј¬¶шЗТАыУГБҪёцЗуәН№жФтјмІйұҫҙОјЖЛг№ШУЪЖҪГжІЁКэј°ІјАпФЁЗшkөгөДСЎИЎКЗ·сКХБІЎЈөЪТ»ёц№жФтТӘЗуІЈ¶ч
УРР§өзәЙЦ®әНОӘБгЈ¬јҙ Ј»өЪ¶юёц№жФтТӘЗуЛщУРФӯЧУФЪҫщәвЖҪТЖМхјюПВҫЯУРКЬБҰІ»ұдРФЈ¬јҙ
Ј»өЪ¶юёц№жФтТӘЗуЛщУРФӯЧУФЪҫщәвЖҪТЖМхјюПВҫЯУРКЬБҰІ»ұдРФЈ¬јҙ ЎЈУЙУЪЙБРҝҝуҪб№№ҫЯУРБў·Ҫ¶ФіЖРФЈ¬¶ФТ»ёш¶ЁФӯЧУЈ¬ҪйөзҫШХуәНІЈ¶чУРР§өзәЙНкИ«¶ФҪЗ»ҜЈ¬јҙСШxЎўyәНzЦб·ЦұрҫЯУРН¬СщөДЦөЎЈұИҪПұн1ҝЙЦӘЈ¬AlNЎўAlPәНAlAsөДҪйөзіЈКэҫщіКФцҙуЗчКЖЈ¬ХвЦчТӘКЗУЙУЪЖдҙшП¶ЦрІҪұдХӯәН·СГЬДЬПВҪөЈ¬К№өГјЫҙш¶ҘөДөзЧУИЭТЧұ»јӨ·ўЈ¬ІъЙъј«»ҜЈ¬ТтҙЛЈ¬ҪйөзіЈКэТАҙОФцјУЎЈ
ЎЈУЙУЪЙБРҝҝуҪб№№ҫЯУРБў·Ҫ¶ФіЖРФЈ¬¶ФТ»ёш¶ЁФӯЧУЈ¬ҪйөзҫШХуәНІЈ¶чУРР§өзәЙНкИ«¶ФҪЗ»ҜЈ¬јҙСШxЎўyәНzЦб·ЦұрҫЯУРН¬СщөДЦөЎЈұИҪПұн1ҝЙЦӘЈ¬AlNЎўAlPәНAlAsөДҪйөзіЈКэҫщіКФцҙуЗчКЖЈ¬ХвЦчТӘКЗУЙУЪЖдҙшП¶ЦрІҪұдХӯәН·СГЬДЬПВҪөЈ¬К№өГјЫҙш¶ҘөДөзЧУИЭТЧұ»јӨ·ўЈ¬ІъЙъј«»ҜЈ¬ТтҙЛЈ¬ҪйөзіЈКэТАҙОФцјУЎЈ
ұн1 КөСйәНАнВЫјЖЛгөДІЈ¶чУРР§өзәЙәНёЯЖөҪйөзіЈКэ
Table 1 Calculated and experimental Born effective charge and dielectric constant

УГұҫҙОөДјЖЛгЦөУлЖдЛьКөСйЦөұИҪП·ўПЦЈ¬ІЈ¶чУРР§өзәЙұ»өН№А¶шҪйөзіЈКэұ»ёЯ№АЈ¬ХвЦчТӘКЗУЙУЪұҫҙОјЖЛгІЙУГөДКЗTMШНКЖЈ¬Г»УРҝјВЗәЛөзЧУ¶ФІЈ¶чУРР§өзәЙөД№ұПЧЎЈБнНвЈ¬LDAФЪҙҰАн°лөјМеІДБПКұЈ¬НЁіЈ»бөН№Аҫ§ёсіЈКэЈ¬К№өГЙщЧУөДұҫХчЖөВКМбёЯЈ¬КҪ(2)ЦРөДөЪ¶юПоФцјУЎЈОӘБЛЛөГчLDA¶ФҪйөзіЈКэөДУ°ПмЈ¬ККөұФцјУҫ§ёсіЈКэ1%Ј¬ІўІЙУГ№гТеМЭ¶ИҪьЛЖ(GGA)¶ФІЈ¶чУРР§өзәЙәНҪйөзіЈКэҪшРРЦШЛгЎЈҪб№ыПФКҫЈ¬LDA¶ФІЈ¶чУРР§өзәЙјёәхГ»УРУ°ПмЈ¬ө«¶ФҪйөзіЈКэөДУ°ПмұИҪППФЦшЈ¬ЖдЦө·ЦұрОӘ4.52Ј¬7.97әН9.73Ј¬¶ФAlPәНAlAsөДУ°ПмјёәхҙпөҪ4%ЎЈұИҪПұҫҙО№ӨЧчөДјЖЛгЦөУлЖдЛьОДПЧ[20?22]өДАнВЫЦө·ўПЦЈ¬Т»ЦВРФҪПәГЎЈ
2.3 ІДБПөДөҜРФРФЦК
ПЯРФөҜРФіЈКэНЁіЈ¶ЁТеОӘЛщКЬУҰБҰУлЛщТэЖрөДУҰұдЦ®Оў·ЦЈ¬јҙ ЎЈЛДҪЧөҜРФХЕБҝТ»°гУөУР
ЎЈЛДҪЧөҜРФХЕБҝТ»°гУөУР
21ёц¶АБўөДХЕБҝФӘЈ¬УЙУЪЙБРҝҝуҪб№№AlNЎўAlPәНAlAsКфУЪБў·Ҫҫ§ПөЈ¬өҜРФХЕБҝјхЙЩОӘ3ёц¶АБўөДХЕБҝФӘ
C11Ј¬C12әНC44ЎЈЙБРҝҝуҪб№№өҜРФіЈКэ Ј¬
Ј¬ Ј¬ө«C44өДјЖЛгІ»ИзC11әНC12өДјЖЛгДЗСщјтөҘЎЈИз№ыУРТ»ёцСШ[111]Цб·ҪПтөДЕтХНЈ¬ФЪҫ§°ыДЪФӯЧУөДО»ЦГІ»ФЩУЙ¶ФіЖРФЛщҫц¶ЁЈ¬¶шұШРлХТөҪЧчУГФЪФӯЧУЙПөДБҰОӘБгөДО»ЦГЈ¬ХвСщЛЙіЪУлОҙЛЙіЪөДC44І»ФЩПаөИЈ¬ЛьГЗВъЧгNielsenәНMartin№ШПөКҪ[23]Ј¬јҙ
Ј¬ө«C44өДјЖЛгІ»ИзC11әНC12өДјЖЛгДЗСщјтөҘЎЈИз№ыУРТ»ёцСШ[111]Цб·ҪПтөДЕтХНЈ¬ФЪҫ§°ыДЪФӯЧУөДО»ЦГІ»ФЩУЙ¶ФіЖРФЛщҫц¶ЁЈ¬¶шұШРлХТөҪЧчУГФЪФӯЧУЙПөДБҰОӘБгөДО»ЦГЈ¬ХвСщЛЙіЪУлОҙЛЙіЪөДC44І»ФЩПаөИЈ¬ЛьГЗВъЧгNielsenәНMartin№ШПөКҪ[23]Ј¬јҙ

УЙУЪЗ°¶јұИҪП·ыәПХжКөПөНіЈ¬ұҫОДЧчХЯЦШөгМЦВЫБЛЛЙіЪХЕБҝЈ¬ІўФЪәуГжөДјЖЛгЦРҫщК№УГЛЙіЪөҜРФХЕБҝЎЈ
јЖЛгөДөҜРФіЈКэәНЖдЛьОДПЧөДАнВЫәНКөСйЦөБРіцФЪұн2ЦРЎЈҫНұҫОДЧчХЯЛщЦӘЈ¬ҪцУРAlAsөДКөСйөҜРФіЈКэҝЙТФАыУГЈ¬ұИҪПұҫОДөДјЖЛгЦөУлОДПЧ[24]өДІјАпФЁЙўЙдІвБҝЦөЈ¬ұҫјЖЛгЦөөНУЪКөСйЦөЈ¬ХвЦчТӘУлұҫҙОјЖЛгШНКЖөДСЎИЎУР№ШЈ¬Troullier-MartinsШНКЖКЗПаөұИнөДЈ¬ТтОӘ°ләЛөзЧУГ»УРұ»ГчИ·өДҙҰАнЈ¬¶шКЗҪ«Ль¶іҪбФЪАлЧУәЛДЪЈ¬ОӘБЛ¶ЁБҝЛөГчШНКЖ¶ФұҫҙОјЖЛгөДУ°ПмЈ¬
ұҫОДЧчХЯІЙУГHartwigsen-Goedeker-HutterШНКЖәуЦШРВјЖЛгөҜРФіЈКэЈ¬ХвёцШНКЖҪ«AsөД3d°ләЛөзЧУЧчОӘјЫөзЧУҙҰАнЈ¬ЛщТӘЗуөДКХБІұИІЙУГTroullier-MartinsШНКЖА§ДСөГ¶аЈ¬ЖҪГжІЁ¶ҜДЬҪШ¶ПДЬҙпөҪ80 HartreeәуЧЬДЬәНБҰІЕКХБІЈ¬ІЙУГХвЦЦШНКЖәуAlAsөДөҜРФіЈКэТАҙООӘ110.6Ўў53.8әН53.3ЎЈ
ҙЛНвЈ¬ІјАпФЁЗшөДMPНшёс»®·ЦГЬ¶ИТІ¶ФөҜРФіЈКэјЖЛгҫ«¶ИУРТ»¶ЁөДУ°ПмЈ¬өұІјАпФЁЗшөДMPНшёс»®·ЦГЬ¶ИФцҙуОӘ16ЎБ16ЎБ16Ј¬өҜРФіЈКэМбёЯ1%ЧуУТЎЈ
ұИҪПұҫҙОјЖЛгөДAlNЎўAlPәНAlAsөДөҜРФіЈКэҝЙЦӘЈ¬AlNөДөҜРФіЈКэГчПФёЯУЪAlPәНAlAsөДөҜРФіЈКэЈ¬¶шAlPәНAlAsПаІоІ»ҙуЎЈЖдФӯТтКЗУЙУЪЛжЧЕТхАлЧУФӯЧУРтКэөДФцјУЈ¬Жд»ҜәПОпөДҫ§ёсіЈКэТІЛжЦ®ФцјУЈ¬¶шТхАлЧУУлСфАлЧУЦ®јдөДөзёәРФЦ®ІојхРЎЈ¬АлЧУУлАлЧУЦ®јдөДҪбәПБҰПВҪөЈ¬өјЦВ3ЦЦ»ҜәПОпөДөҜРФіЈКэТАҙОПВҪөЎЈУЦУЙУЪAlNөДҫ§ёсіЈКэГчПФөДРЎУЪAlPәНAlAsөДҫ§ёсіЈКэЈ¬ө«AlPәНAlAsөДҫ§ёсіЈКэПаІоОЮјёЈ¬јЫөзәЙГЬ¶И·ЦІјПаІоІ»ҙу(јыНј2)ЎЈФЩХЯЈ¬ФЪPәНAsШНКЖөДСЎФсЦРГ»УРҝјВЗДЪәЛөзәЙөДІоТмЈ¬ТтҙЛЈ¬AlNөДөҜРФіЈКэГчПФёЯУЪAlPәНAlAsөДөҜРФіЈКэЈ¬¶шәуБҪХЯөДЦөПаІоІ»ҙуЎЈ
ОӘБЛёьәГөШЛөГчХвР©ҫ§МеөДөҜРФРРОӘЈ¬КҫТвөШ»ӯіцХвР©ІДБПөДөҜРФДЈБҝұнХчГж(јыНј3)ЎЈҫЎ№ЬЙБРҝҝуҪб№№ҫЯУРёЯ¶ФіЖРФЈ¬ө«І»ДЬУГТ»ёцөҘТ»өДГжАҙұнКҫХыёцҫ§МеөДөҜРФРРОӘЈ¬КөјКЦР·ЗіЈУРУГөДТ»ёцГжКЗұнКҫІ»Н¬·ҪПтөҜРФДЈБҝұд»ҜөДГжЎЈФЪҙЛЈ¬јЖЛг3ЦЦ»ҜәПОп°лөјМе(101)ГжөДөҜРФДЈБҝЈ¬ІўАыУГПВГ湫КҪҪшРРјЖЛг[25]

ҙУНјЦРҝЙТФҝҙіцБў·Ҫҫ§ПөЦРөҜРФДЈБҝТІІ»КЗёчПтН¬РФЎЈЛж·ҪПтөДұд»ҜИЎҫцУЪ Ј¬ХвёцБҝФЪЁў100?·ҪПтОӘБгЈ¬ФЪЁў111?·ҪПтҙпөҪЧоҙуЦө1/3ЎЈКёҫ¶ЦұҪУХэұИУЪөҜРФДЈБҝөДГжЈ¬КЗТ»ёцФЪёчГжЦРСл°јПЭөДФІҪЗБў·ҪМеЎЈөҪДҝЗ°ОӘЦ№Ј¬»№ОҙјыУРХвР©ІДБПМШХчГжөҜРФДЈБҝөДјЖЛгұЁөАЎЈ
Ј¬ХвёцБҝФЪЁў100?·ҪПтОӘБгЈ¬ФЪЁў111?·ҪПтҙпөҪЧоҙуЦө1/3ЎЈКёҫ¶ЦұҪУХэұИУЪөҜРФДЈБҝөДГжЈ¬КЗТ»ёцФЪёчГжЦРСл°јПЭөДФІҪЗБў·ҪМеЎЈөҪДҝЗ°ОӘЦ№Ј¬»№ОҙјыУРХвР©ІДБПМШХчГжөҜРФДЈБҝөДјЖЛгұЁөАЎЈ
ұн2 јЖЛгөДЛЙіЪАлЧУөҜРФіЈКэ
Table 2 Calculated relaxed-ion elastic constant


Нј3 ЙБРҝҝуҪб№№AlN, AlPәНAlAsМШХчГж(101)ГжөДөҜРФДЈБҝ
Fig.3 Calculated elastic moduli of characteristic plane (101) for zinc-blende AlN, AlP and AlAs
3 ҪбВЫ
1) ІЙУГГЬ¶ИәҜКэАнВЫПВөДҫЦУтГЬ¶ИҪьЛЖЈ¬ЙБРҝҝуҪб№№AlNЎўAlPәНAlAsөДҫ§ёсІОКэУЕ»ҜЦөВФРЎУЪКөСйЦөЈ¬ХвЦчТӘКЗҫЦУтГЬ¶ИҪьЛЖөДҫЦПЮЛщЦВЈ¬ІўҪш¶шУ°ПмөҪАнВЫҙшП¶ЦөЎЈ
2) »щУЪјЖЛгөДҫ§ёсІОКэУЕ»ҜЦөЈ¬јЖЛгЛщөГөДХвР©ІДБПөДІЈ¶чУРР§өзәЙәНҪйөзіЈКэУлКөСйЦөұИҪП·Цұрұ»өН№АәНёЯ№АЈ¬ЦчТӘУлұҫҙОјЖЛгЛщІЙУГөДШНКЖәНҪ»»»№ШБӘКЖУР№ШЎЈ
3) »щУЪјЖЛгөДҫ§ёсІОКэУЕ»ҜЦөЈ¬ІЙУГГЬ¶ИәҜКэИЕ¶ҜАнВЫјЖЛгБЛХвР©ІДБПөДөҜРФДЈБҝЈ¬УлЖдЛьІОҝјОДПЧМṩөДАнВЫјЖЛгЦөТ»ЦВРФҪПәГЈ¬ө«ІЙУГІ»Н¬өДјЖЛгДЈРНәНШНКЖ»бТэЖрОўРЎөДІоТмЎЈ
4) АыУГјЖЛгЛщөГөДөҜРФіЈКэЈ¬»жЦЖБЛХвР©ІДБПМШХчЖҪГж{101}өДөҜРФДЈБҝЖҪГжНјЈ¬ҫЎ№Ь»№Г»УРКөСйәНАнВЫЦөЧчұИҪПЈ¬ө«ОӘҪсәу¶ФХвР©ІДБПөДБҰС§СРҫҝМṩҪијшЎЈ
REFERENCES
[1] WANG H Y, XU H, XIAO J R, LI M J. Electronic structure, dielectric and dynamical properties of zinc-blended AlN from first principles calculation[J]. Mod Phys Lett B, 2007, 21: 1775?1784.
[2] әВПюҫІ, өіЦЗГф, РмәЈЖј. ёЯөјИИВКј°өНҪйөзіЈКэөДAlN/PIДЙГЧёҙәПұЎДӨСРҫҝ[J]. №ҰДЬІДБП, 2007, 38(10): 1618?1620.
HAO Xiao-jing, DANG Zhi-min, XU Hai-ping. Research on aluminum nitride/polyimide nanocomposite films with high thermal conductivity and low dielectric permittivity[J]. Journal of Functional Materials, 2007, 38(10): 1618?1620.
[3] ХЕІДИЩ, іВәкЙЖ, іВУсәм. Al8P8НЕҙШ»·ЧҙҪб№№УлРФЦКөДГЬ¶И·әәҜАнВЫСРҫҝ[J]. ФӯЧУУл·ЦЧУОпАнС§ұЁ, 2006, 21(12): 1368?1372.
ZHANG Cai-rong, XU Guang-ji, CHEN Hong-shan, CHEN Yu-hong, LI Wei-xue. Density functional theory study on the structure and properties of Al8P8 clusters[J]. Acta Phys Chim, 2005, 21(12): 1368?1372.
[4] WANG X, VANDERBILT D. First-principles perturbation of dielectric and Born charge tensors in finite electric fields[J]. Phys Rev B, 2007, 75: 115116?115121.
[5] Вн Бъ, ХЕ Со, ҙч Сп, Соё»»Ә. ёЯРФДЬInGaAs/AlAs№ІХсЛнҙ©¶юј«№ЬөДСРЦЖУлЖчјюДЈДв·ЦОц[J]. °лөјМеС§ұЁ, 2007, 28(4): 563?566.
MA Long, ZHANG Yang, DAI Yang, YANG Fu-hua. Fabrication and device simulation of high performance InGaAs/AlAs resonant tunneling diodes on InP substrates[J]. Chinese Journal of Semiconductors, 2007, 28(4): 563?566.
[6] РЬ Сж, ёөХэТе, Нх р©. CaF2 ЦъјБ·ЕөзөИАлЧУЙХҪбНёГчAlNМХҙЙөДОў№ЫҪб№№әН№вС§РФДЬ[J]. ЦР№ъУРЙ«ҪрКфС§ұЁ, 2005, 15(11): 1705?1709.
XIONG Yan, FU Zheng-yi, WANG Hao. Microstructure and optical property of transparent AlN ceramics by spark plasma sintering with CaF2[J]. The Chinese Journal of Nonferrous Metals, 2005, 15(11): 1705?1709.
[7] HAMANN D R, WU X, RABE K M, VANDERBILT D. Metric tensors formulation of strain in density-functional perturbation theory[J]. Phys Rev B, 2005, 71: 035117?035129.
[8] WU X, VANDERBILT D, HAMANN D R. Systematic treatment of displacements, and electric fields in density-functional perturbation theory[J]. Phys Rev B, 2005, 72: 035105?035117.
[9] PERDEW J P, ZUNGER A. Self-interaction correction to density-functional approximation for many-electron systems[J]. Phys Rev B, 1991, 23: 5048?5079.
[10] TROULLIER N, MARTINS J L. Effective pseudopotentials for plane-wave caculations[J]. Phys Rev B, 1991, 43: 1991?2006.
[11] HENDRIK J, MONKHORST, PACK J D. Special points for Brillouin-zone intergrations[J]. Phys Rev B, 1976, 13: 5188?5192.
[12] PETROV I, MOJAB E, POWELL R C, GREEN J E, HULTMAN L, SUNDGREN J E. Synthesis of metastable epitaxial zhc-blende-structure AlN by solid-state reaction[J]. Appl Phys Lett, 1992, 60: 2491?2493.
[13] LUCOVSKY G, MARTIN R M, BURSTEIN E. Localized effective charges in diatomic crystals[J]. Phys Rev B, 1971, 4: 1367?1374.
[14] CORSO A D, MAURI F, RUBIO A. Density-functional theory of the nonlinear optical susceptibility: Application to cubic semiconductors[J]. Phys Rev B, 1996, 53: 15638?15642.
[15] MADELUNG O, SCHULTZ M, WEISS H. Physics of group ўф elements and ўу-ўх compounds, of Landolt-B?rnstein, numerical data and functional relationships in science and technology[M]. New York: Springer, 1982: 17?44.
[16] SHANG G, PEACOCK P W, ROBERTSON J. Stability and band offsets of nitrogenated high-dielectric-constant gate oxides[J]. Appl Phys Lett, 2004, 84: 106?108.
[17] GONZE X, LEE C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory[J]. Phys Rev B, 1997, 55: 10355?10368.
[18] The ABINIT code is a common project of the UniversitЁҰ Catholique de Louvin, Corning Incorporated, and other contributors, URL http://www.abinit.org
[19] GOLDBERG Y. Properties advanced semiconductor materials GaN, AlN, BN, SiC, SiGe[M]. New York: Wiley, 2001: 31?47.
[20] SHIMADA K, SOTA T, SUZUKI K. First-principles study on electronic and elastic properties of BN, AlN, and GaN[J]. J Appl Phys, 1998, 84: 4951?4958.
[21] KARCH K, BECHSTEDT F. Ab initio lattice dynamics of BN and AlN: Covalent versus ionic forces[J]. Phys Rev B, 1997, 56: 7404?7415.
[22] BERNARDINI F, FIORENTINI V. Electronic dielectric constants of insulators calculated by the polarization method[J]. Phys Rev B, 1998, 58: 15292?15295.
[23] NIELSON O H, MARTIN R M. First-principles calculation of stress[J]. Phys Rev Lett, 1983, 50: 697?700.
[24] KRIEGER M, SIGG H. Elastic constants and Poisson ratio in the system AlAs-GaAs[J]. Appl Phys Lett, 1995, 66: 682?684.
[25] NYE J F. Physical properties of crystals their representation by tensors and matrices[M]. Oxford: Clarendon, 1985: 88?101.
»щҪрПоДҝЈә№ъјТЧФИ»ҝЖС§»щҪрЧКЦъПоДҝ(50271085)
КХёеИХЖЪЈә2007-10-20Ј»РЮ¶©ИХЖЪЈә2008-05-16
НЁС¶ЧчХЯЈәНх»АУСЈ¬І©КҝСРҫҝЙъЈ»өз»°Јә13975742740Ј»E-mail: whycs@163.com
(ұајӯ Бъ»іЦР)

