中国有色金属学报 2004,(07),1106-1111 DOI:10.19476/j.ysxb.1004.0609.2004.07.009
粉末冶金发泡时泡沫铝孔结构及泡壁的微观组织演变
朱勇刚 陈锋 梁晓军 何德坪
摘 要:
研究了粉末冶金法制备泡沫铝时泡沫孔结构及胞壁微观组织变化的规律。泡沫铝发泡时经历微膨胀、显著膨胀和收缩等过程;孔结构经历了形核和长大,以及在毛细力和重力驱动下泡壁熔体的流动引起的合并粗化和孔隙率自上而下梯度减小等演化过程。加热时,铝/硅颗粒边界处硅的扩散层首先熔化,并沿着铝颗粒边界扩散,最终使铝硅粉末复合体完全熔化而实现合金化。实验发现泡壁的凝固组织与典型的变质处理后的铸造铝硅合金的组织类似。
关键词:
泡沫铝 ;粉末冶金 ;孔结构 ;微观组织 ;
中图分类号: TF124
作者简介: 朱勇刚(1977),男,硕士研究生.; 陈 锋,教授,博士;电话:02583794119;E mail:fengchen@seu.edu.cn;
收稿日期: 2003-10-27
基金: 国家自然科学基金资助项目(50231010);
Evolution of pore morphology and cell wall microstructure of aluminum foam during powder metallurgical foaming
Abstract:
Evolution of pore morphology and cell wall microstructure of aluminum foam during powder metallurgical foaming was studied. It was found that aluminum foam shows three stages during foaming, that is tiny expansion, remarkable expansion and shrinkage; and the pore morphology goes through three stages, that is nucleation and growth, merging and coarsening, and the porosity decreasing from the upper part of the foam to the lower part of the foam. The merging and porosity gradient of the pore structures were resulted from capillarity-driven and gravity-driven melt flow within the melt foam column. The silicon diffusing layer along the boundary of aluminum and silicon powders melts firstly during the heating, and then the melt diffuses along the boundary of the aluminum powders, resulting in the entire alloying between aluminum and silicon powders. The experiment shows that the solidified cell wall microstructures are similar to those of typical modified cast Al-Si alloys.
Keyword:
aluminum foam; powder metallurgy; pore morphology; microstructure;
Received: 2003-10-27
泡沫金属是一种应用前景广阔的新型结构与功能兼容材料。 作为结构材料, 具有轻质、 高比强度的特点; 作为功能材料, 具备隔声(或吸声)、 隔热(或散热)、 阻燃、 阻尼、 冲击能量吸收和电磁屏蔽等多种物理性能
[1 ,2 ,3 ,4 ,5 ]
。 目前已有多种制备泡沫金属的方法
[1 ,2 ,3 ,4 ]
, 其中粉末冶金发泡法
[6 ,7 ,8 ,9 ,10 ,11 ]
由于能够生产复杂形状的泡沫铝及泡沫铝芯三明治结构而越来越受到研究者的重视。 1999年起德国DFG(德意志研究联合会)设立"胞状金属"的优先研究计划, 联合20多个著名高校和国立研究所从事粉末冶金法为主的胞状金属制备和性能研究, 应用目标为汽车工业。 奥地利和斯洛伐克等欧洲高校和公司也有相应的研究。 在国内, 粉末冶金发泡法则刚起步不久, 鲜有研究报道。 目前, 国外这一相关研究主要集中在性能上, 制备工艺涉及较少。 此外, 粉末冶金发泡时往往采用元素粉而不采用预合金粉配制, 其优点是可方便地改变合金成分, 但元素粉在发泡过程中(加热温度较低, 时间较短)如何实现合金化的机制迄今未见报道。
本文作者采用自行研制的设备, 用粉末冶金发泡法制备了Al-Si泡沫合金, 并对发泡过程中孔结构的演变及铝、 硅元素粉的合金化过程进行了探索。
1 实验
实验采用的合金成分(质量分数)为Al-7%Si(AlSi7)。 将铝粉(纯度99%)、 硅粉(纯度99.9%)、 发泡剂(TiH2 粉, 纯度99%)按比例混合。 本实验TiH2 的含量固定为1%(质量分数)。 将3种粉末在混粉机中混合均匀, 然后在模具中进行轴向冷压和热压, 获得致密度98%以上的可发泡预制件, 尺寸为d 45 mm×7.5 mm。
将预制件放入圆柱型的钢模中, 再置于640 ℃的电阻炉中发泡, 并用热电偶测量预制件的温度变化规律。 将不同加热时间的预制件从炉中取出, 迅速冷却, 测量其等效膨胀高度, 并观察其截面孔结构。 将试样镶嵌、 抛光和浸蚀, 用光学显微镜和扫描电镜(SEM)观察泡壁的微观组织变化, 并用X射线能谱仪(EDX)进行微观成分分析。
由于粉末冶金泡沫化过程对TiH2 质量十分敏感, 为此用新研制的气相层析仪对TiH2 的热分解曲线进行测试
[12 ]
。
2 结果与讨论
2.1 TiH2的热分解过程
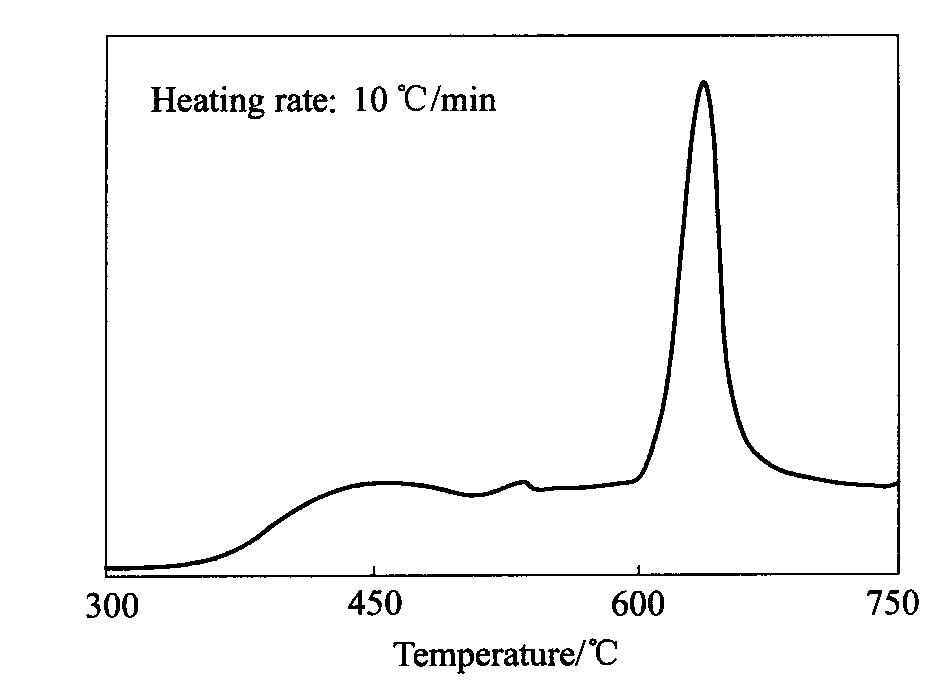
TiH2 的热分解曲线见图1
[12 ]
, 其分解开始温度在400 ℃左右, 600 ℃左右开始大量分解, 640 ℃左右达到峰值。 预制件中的TiH2 处于受压状态, 其分解受到一定的抑制而趋向于在更高温度下分解
[13 ]
。 但图1可为揭示TiH2 在预制件中的分解规律提供参考。
2.2 发泡时孔结构的演化规律
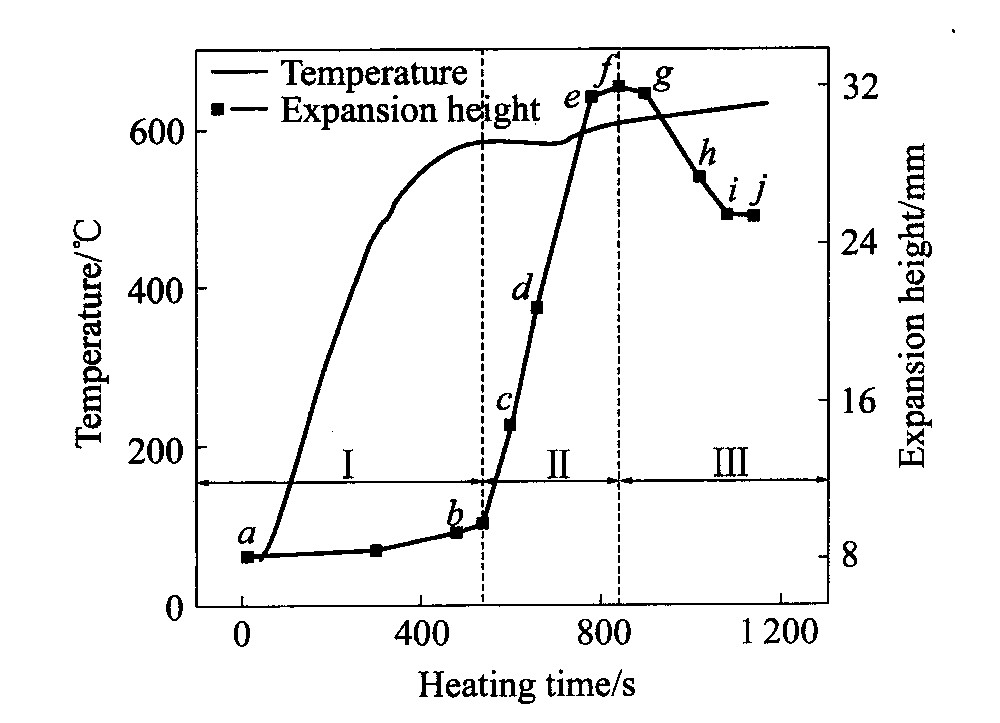
图2所示为预制件的温度和膨胀高度与加热时
图1 TiH2热分解曲线 Fig.1 Thermal decomposition curve of TiH2
图2 AlSi7合金发泡过程中的温度 和膨胀曲线 Fig.2 Temperature and expansion curves of AlSi7 sample during foaming
间的关系曲线。 可见, 预制件升温时, 在582 ℃附近出现平台, 表明熔化开始。 随平台区结束, 预制件完全熔化, 熔体温度继续上升, 并接近640 ℃的平衡温度。 对应于图2所示为预制件膨胀曲线。 可见预制件在固态及刚发生熔化时只产生微小的膨胀(Ⅰ区)。 随着熔化的进行, 预制件发生快速膨胀, 并一直持续到预制件完全熔化后, 直达到膨胀最大值(Ⅱ区)。 随着加热时间的延长, 熔体温度缓慢升高, 预制件又发生收缩, 并最终达到稳定(Ⅲ区)。
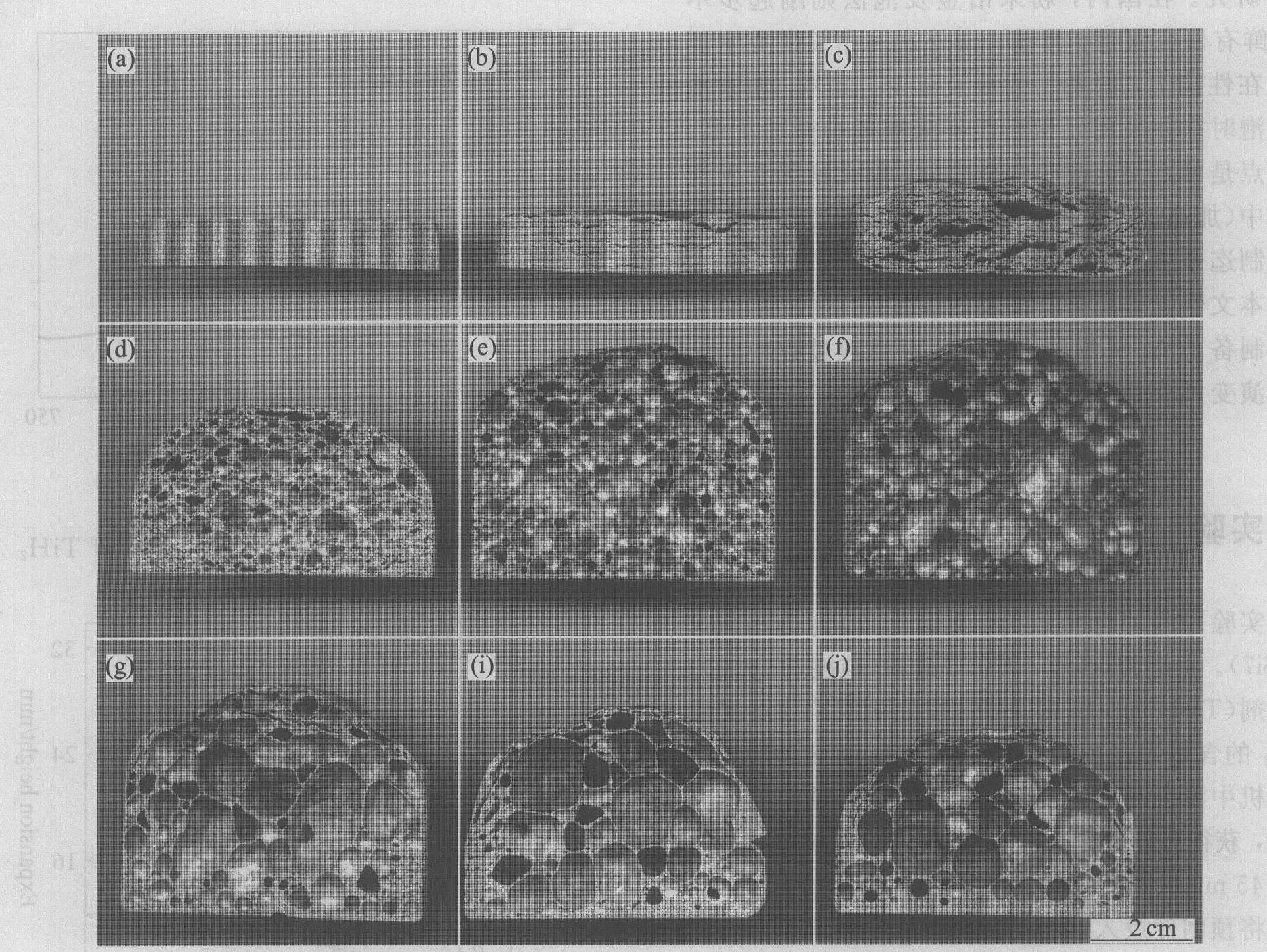
图3所示为与图2中各发泡阶段a ~j 相对应的预制件的剖面图。 由图可以看到发泡过程中孔结构的形成与演化过程。
1) 孔的形成。
在400 ℃之前, 试样完全处于固态, TiH2 几乎未分解(图1), 此时膨胀极微。 随着温度升高, TiH2 开始分解, 而预制件强度下降。 因
图3 不同加热时间的AlSi7合金孔结构演变过程 Fig.3 Evolution of pore morphology of AlSi7 alloy heated for different time (a)―0 s; (b)―480 s; (c)―600 s; (d)―660 s; (e)―780 s; (f)―840 s; (g)―900 s; (i)―1 020 s; (j)―1 080 s
预制件横向密度(强度)较轴向低, 接近熔点时(约580 ℃), TiH2 分解所产生的压力使预制件产生横向裂纹(图3(b))。
2) 孔的长大。
随加热时间延长, 温度升高到582 ℃附近时, 试样开始熔化, 处于液固共存状态, 其粘度显著降低。 当预制件完全熔化以及进一步升温到600 ℃左右时, 粘度进一步降低; 温度处于580~600 ℃之间时, 即TiH2 大量分解的温度, 为预制件的快速膨胀提供了条件。 随着孔隙率的提高, 气孔由裂隙和球状(厚液膜)逐渐发展为多面体状孔(薄液膜), 直至达到最大的膨胀率(图3(c)~3(f))。
3) 孔的破裂和合并。
在最大膨胀之后, 顶部泡沫出现破裂和坍塌; 中部泡沫合并形成大而不规则的气孔; 因胞壁熔体在重力作用下自上而下的流动(即排液现象), 底部泡壁增厚, 并出现实体层, 使泡沫总体孔隙率降低(图3(i), 3(j))。
液态金属泡沫的上述合并和排液现象是与泡壁熔体在毛细作用和重力驱动下的流动相关联的。 泡沫的截面结构可简化为图4所示的模型, 其中泡面
图4 简化的泡面和泡边界结构示意图 Fig.4 Simplified schematic diagram of cell face and plateau border
(Cell face, CF)因曲率半径较大可简化为平面, 其厚度为x CF ; 内部压力为p G , 等于相邻泡面气体的压力。 多个泡面相交的边界(称之为Plateau border, PB)
[14 ]
处的曲率半径为r PB 。 其内部压力设为p PB 。 由于泡壁熔体表面张力的作用, PB和CF处存在压力差, 即
Δp =p PB -p G =-γ /r PB (1)
式中 γ 为熔体的表面张力。
在此压力差的驱动下, CF处的熔体向PB处流动, 使CF减薄。 采用改进的Reynolds方程
[14 ]
, CF的减薄速率为
v CF =dx CF /dt ∝2x
C
F
2
Δ p/3μ (2)
式中 μ为熔体的粘度。
可见CF 的减薄速度与μ成反比, 与Δ p也即γ成正比。 CF 减薄到50~80 μm 时即发生破裂
[15 ]
, 使泡沫合并和粗化。
另一方面, 聚集在PB 处的熔体在重力作用下会沿PB 通道网络向下流动, 其结果是上部PB 处的熔体体积小于下部PB 处的熔体体积。 由方程(1)可知, 上部PB 处的压力小于下部, 即毛细作用力在一定程度上会阻碍熔体的向下流动。 综合重力和毛细力作用结果, 熔体沿PB 通道网络向下流动的平均速度有以下关系
[14 ]
:
vPB d (1/rPB d z)/μ (3)
式中 ρ为熔体密度; g为重力加速度; z为纵向坐标。
可见PB 处的排液与ρ成正比, 而与μ成反比。 排液的结果使泡沫柱中的熔体重新分布, 即上部的孔隙率增大, 下部孔隙率减小, 直至底部形成实体。
熔体泡沫结构演变是泡壁熔体在毛细力和重力驱动下的流动引起的, 该过程从预制件出现液相一直持续到泡沫熔体完全凝固为止。 由上述分析可知, 提高泡沫结构稳定性的关键是使熔体有足够的粘度, 这就要求发泡温度不宜过高(因熔体粘度随温度的增高而降低); 合金较宽的固液两相区(如本文采用的AlSi 7合金)也能减缓这一过程。 更可靠的方法是使熔体中含有一定体积含量的微细陶瓷颗粒以增加粘度
[15 ]
, 该方面的探索本文作者正在进行之中。
液态泡沫在冷却、 凝固过程中结构还会发生一定程度的演变, 因而图3(d )~3(f )阶段孔结构(主要为图3(e )~3(f )阶段, 因温度高于固相线)较高温状态下的实际孔结构滞后。 通过测量冷却时泡沫芯部的温度变化曲线, 发现该滞后约在20 s 左右, 显著小于图3(e )~3(f )阶段的时间间隔, 因而图3(d )~3(f )可以近似描述各加热阶段孔结构的变化规律。 新近发展的X 射线透视法可以实时测量熔体泡沫结构的动态演变过程
[16 ]
, 但得到的图像为沿样品厚度方向孔结构的叠加, 分辨率较差, 因而实时泡沫结构的显示技术还有待进一步发展。
2.3发泡时胞壁微观组织的演化及铝硅合金化的机制
图5所示分别为图2(a)~(d), (f)和(i)各发泡阶段预制件(泡壁)的微观组织。 由于冷却时泡壁中的铝硅合金化过程还会继续, 因而图5(c ), 5(d ), 5(f )揭示的合金化过程较高温下的实际状况有一定的滞后, 但滞后较小, 并不影响铝硅合金化机制的建立。
图5(a ), 5(b )所示分别为未加热和加热480 s 预制件的微观组织。 能谱成分分析表明, 深灰色的颗粒为Si , 淡灰色的为纯Al 。 由于此时预制件未熔化, 硅颗粒仍旧独立地嵌在铝基体中, 因而其微观
图5 不同发泡阶段泡壁的微观组织 Fig.5 Microstructures of AlSi7 foam heated for different time (a)―0 s; (b)―480 s; (c)―600 s; (d)―660 s; (f)―840 s; (i)―1 020 s
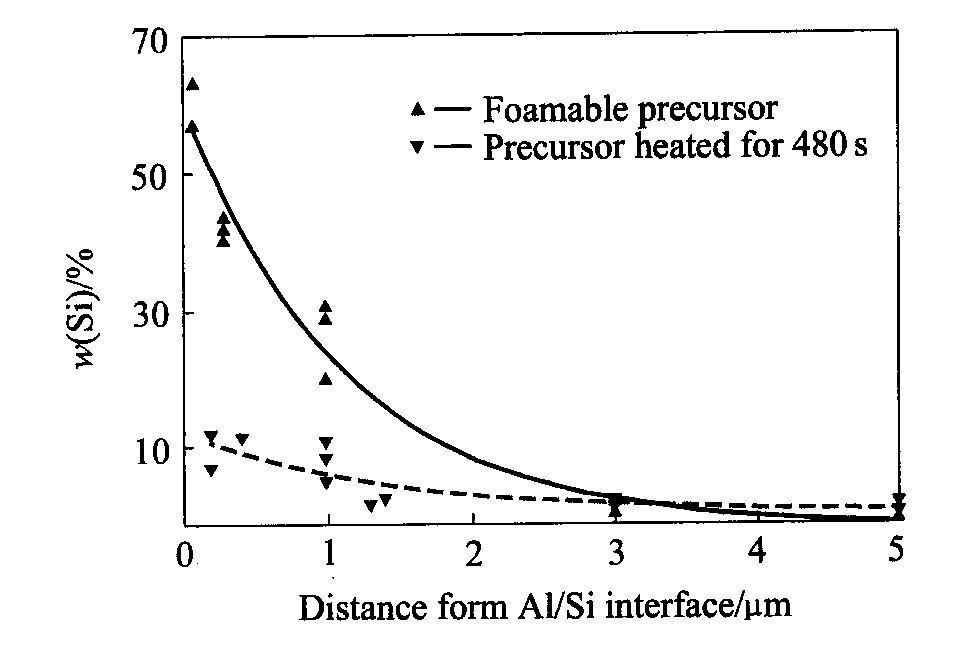
组织相似。 图6所示为图5(a)的高倍扫描电镜照片, 可见铝基体中存在网状裂纹, 而硅颗粒均处于这些裂纹的交界处。 EDX微观成分分析显示, 这些裂隙处氧原子摩尔分数达到1.96%。 据此可以认为, 这些裂隙正是压缩后铝粉颗粒的边界。 EDX分析表明, 在铝/硅界面附近的铝基体中存在着硅的扩散边界层, 宽度约5 μm, 如图7所示。 其中距铝/硅边界3 μm内的硅浓度大大超过平衡条件下硅在铝中的最大溶解度1.59%(摩尔分数)(577 ℃, 1.01×105 Pa)
[17 ]
。
图6 未发泡预制件的SEM像 Fig.6 SEM image of foamable AlSi7 precursor
图5(c), 5(d)所示分别为加热600和660 s预制件(泡壁)的微观组织。 发现硅颗粒细化, 并沿着铝颗粒边界分布。 图5(f), 5(i)分别为加热840和1 020 s预制件(泡壁)的微观组织。 由于此时预制件完全熔化, 因而显示的是AlSi7合金熔体的凝固组织。
对其中淡灰色的区域进行成分分析显示, 硅含量为1.54%, 其余均为铝, 证实这是先共晶α -Al
[18 ]
。 α -Al晶粒间深灰色区域成分显示, 硅含量为17.61%, 这可能是由于凝固速度较快, 形成了非平衡Al-Si共晶凝固组织。 该组织与典型的变质处理后的铸造Al-Si合金组织相似
[18 ]
, 说明可以用元素粉替代合金粉, 在发泡的较短时间内和较低温度下实现合金化。
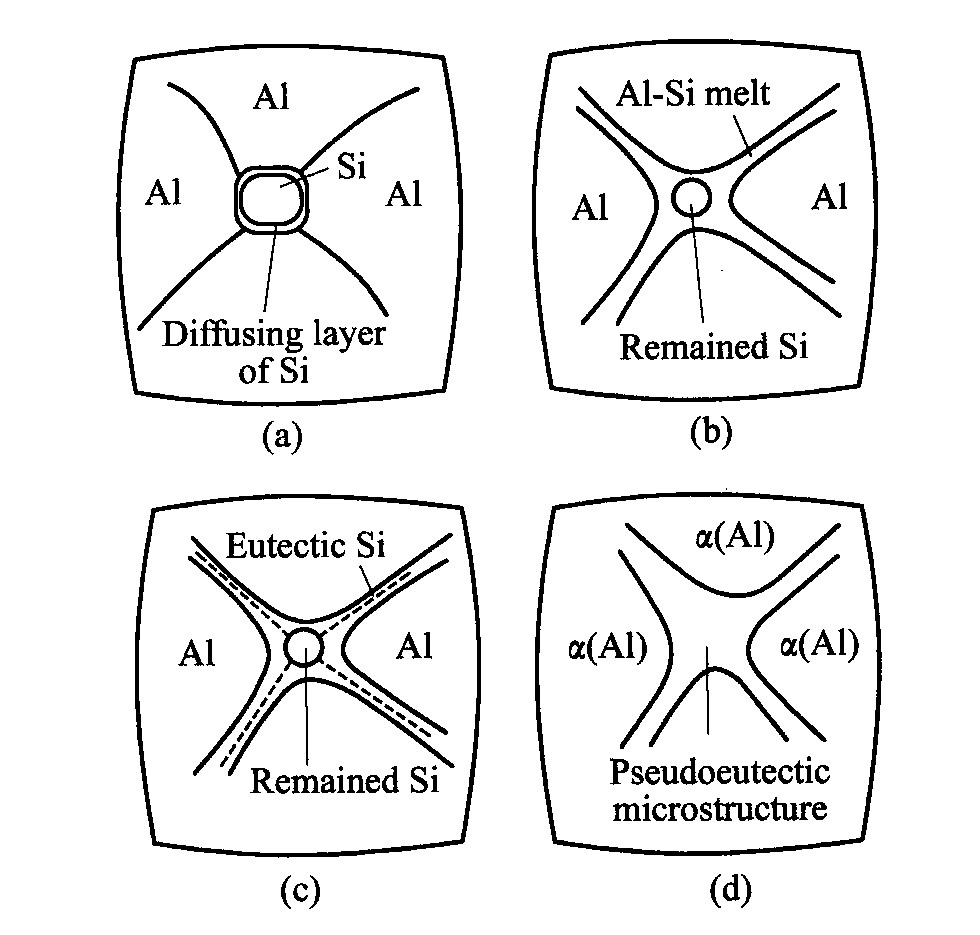
结合图5~7的实验结果, 可提出加热发泡时铝/硅颗粒间实现合金化的模型(如图8所示)。
在450 ℃, 1.01×105 Pa时, Si在铝中的平衡溶解度仅为0.48%(摩尔分数)
[17 ]
。 由于预制件是在400~450 ℃, 250 MPa条件下压制而成, 可能由于超高压的作用, 硅颗粒中硅原子向周围铝基体中扩散, 形成硅原子过饱和扩散边界层(厚度约3 μm)。 在预制件脱模冷却后, 硅原子的这一过饱和状态被保留到室温(见图7, 8(a))。 当预制件加热
图7 铝基体中硅的扩散边界层 Fig.7 Diffusing layer of Si in aluminum matrix
图8 铝/硅颗粒间的合金化模型 Fig.8 Model of alloying process between aluminum and silicon powders (a)―Formable precursor; (b)―Heated to 583 ℃; (c)―Solidified microstructure for Fig.8(b); (d)―Final solidified microstructure
发泡时, 由于压应力的释放及热扩散, 过饱和的硅原子趋向平衡分布, 因而扩散边界层中硅原子的过饱和程度降低, 厚度增加(见图7)。 当预制件升温到582 ℃时, 硅扩散层熔化(低于纯铝的熔点660 ℃), 形成Al-Si熔体层, 并沿着铝颗粒边界扩散(见图8(b))。 此时将预制件取出冷却, Al-Si熔体层的凝固组织将是先共晶α -Al和(α -Al+Si)共晶组织。 其中α -Al附着在Al颗粒上生长, 使共晶Si呈网状沿铝颗粒边界分布(见图5(c), 5(d), 图8(c))。 随着加热的继续, 在有液相存在的情况下, 铝、 硅颗粒更易熔化, 最后在显著低于纯铝的熔点下熔融, 实现合金化。 其凝固组织示意图见图8(d), 实际凝固组织见图5(f), 5(i)。
3 结论
1) 泡沫铝发泡时经历微膨胀、 显著膨胀和收缩等过程; 孔结构经历形核和长大以及毛细力和重力驱动下泡壁熔体流动引起的合并粗化和孔隙率自上而下梯度减小等演化过程。 要提高孔结构的稳定性, 关键是提高熔体的粘度。
2) 加热时, 铝/硅颗粒边界处硅的扩散层首先熔化, 并沿着铝颗粒边界扩散, 使合金快速熔化。 表明由铝和硅元素粉构成的泡壁组织能在发泡的较短时间内和较低温度下实现合金化, 并得到均匀的微观组织。
参考文献
[1] 吴照金,何德坪.泡沫铝凝固过程中孔隙率的变化[J].科学通报,2000,45(8):829835.WUZhao jin,HeDe ping.ChangesinporosityoffoamedAlduringsolidification[J].ChineseScienceBulletin,2000,45(8):829835.
[2] 杨东辉,何德坪.多孔铝合金的孔隙率[J].中国科学(B),2001,31(3):265271.YANGDong hui,HEDe ping.PorosityofporousAlalloy[J].ScienceinChina(B),2001,31(3):265271.
[3] 郑明军,何德坪,戴 戈.胞状铝合金凝固过程中固―液两相区的附加力场[J].中国科学(B),2003,32(5):472480.ZHENGMing jun,HEDe ping,DAIGe.AdditionalforcefieldincoolingprocessofcellularAlalloy[J].ScienceinChina(B),2003,32(5):472480.
[4] BanhartJ.Manufacture,characterizationandapplicationofcellularmetalsandmetalfoams[J].ProgressinMaterialsScience,2001,46:559632.
[5] AshbyMF,EvansAG,FleckNA,etal.MetalFoams:ADesignGuide[M].London:Butterworth―HeinemannPress,2000.610.
[6] KrisztB,CekanP,FaureK.FoamabilityoftheAl Sisystem[A].BanhartJ,AshbyMF,FleckNA.CellularMetalsandMetalFoamingTechnology[C].Bremen:MITVerlag,2001.7782.
[7] BaumgartnerF,GersH.IndustrializationofP/Mfoamprocess[A].BanhartJ,AshbyMF,FleckNA.MetalFoamsandPorousMetalStructure[C].Bremen:MITVerlag,1999.7378.
[8] StanzickH,WichmannM,BanhartJ.Processcontrolinaluminumfoamproductionusereal timeX rayradioscopy[J].AdvancedEngineeringMaterials,2002,4(10):814823.
[9] BanhartJ,StanzickH.Metalfoamevolutionstudiedbysynchrotronradioscopy[J].AppliedPhysicsLetters,2001,78(8):11521154.
[10] BaumeisterJ,BabhartJ,WeberM.Aluminumfoamsfortransportindustry[J].Materials&Design,1997,18(4):217220.
[11] DuarteI,WeigandP,BanhartJ.Foamingkineticsofaluminumalloys[A].BanhartJ,AshbyMF,FleckNA.MetalFoamsandPorousMetalStructure[C].Bremen:MITVerlag,1999.97104.
[12] 杨东辉,何德坪,杨上闰.氢化钛热分解动力学和铝合金泡沫化研究[J].中国科学(B),2004,34(3):195201.YANGDong hui,HEDe ping,YANGShang run.KineticofTiH2decompositionandresearchonmoltenAlalloy sfoamin[J].ScienceinChina(B),2004,34(3):195201.
[13] DuarteI,BanhartJ.AStudyofaluminumfoamformationkineticsandmicrostructure[J].ActaMater,2000,48:23492362.
[14] GergelyV,CurranDC,ClyneTW.Drainageofmetallicfoams:modelingandexperimentalobservations[A].BanhartJ,AshbyMF,FleckNA.MetalFoamsandPorousMetalStructure[C].Bremen:MITVerlag,2001.105108.
[15] BabsanN,LeitlmeierD.Foamabilityofparticlereinforcedaluminiummelt[J].Mat wiss.u.Werksofftech,2003,34:2229.
[16] StanzickH,BanhartJ.Processcontrolinaluminiumfoamproductionusingreal timeX rayradioscopy[J].AdvancedEngineeringMaterials,2002,4(10):814823.
[17] HansenDM.ConstitutionofBinaryAlloys[M].NewYork:TheMaplePressCompany,1958.133.
[18] MondolfoLF.AluminumAlloys:StructureandProperties[M].London:ButterworthsPress,1977.72183