文章编号:1004-0609(2011)07-1668-07
Al、Ti掺杂对Mg2Ni合金相结构稳定性的
影响及其微观机理
张 健1, 黄雅妮2, 华熳煜1, 邵毅敏1, 周惦武3, 彭 平2
(1. 长沙理工大学 汽车与机械工程学院,长沙 410114;
2. 湖南大学 材料科学与工程学院,长沙410082;
3. 湖南大学 汽车车身先进设计制造国家重点实验室,长沙 410082)
摘 要: 采用第一性原理赝势平面波方法,研究元素Al和Ti掺杂对Mg2Ni储氢合金相结构稳定性的影响及其微观机理。结果显示:在掺杂浓度x=0~0.5范围内,所形成的Mg2Ni型Mg2-xMxNi(M=Al, Ti)固溶体合金的相结构稳定性随Al掺杂浓度的增大而增强,随Ti掺杂浓度的增大而减弱,且Mg2-xMxNi(M=Al, Ti)固溶体合金相对于立方结构的Mg3MNi2(M=Al, Ti)化合物呈现热力学不稳定性,极易分解成由立方结构Mg3MNi2(M=Al, Ti)和六方结构Mg2Ni组成的复合相,计算结果与实验结果吻合。电子结构分析表明,Al、Ti掺杂Mg2Ni储氢合金的相结构稳定性与体系在低能级区的成键电子数密切相关。
关键词:Mg2Ni;储氢合金;相结构稳定性;赝势平面波;电子结构
中图分类号:TG139.7 文献标志码:A
Influence and micromechanism of Al or Ti doping on
phase structural stability of Mg2Ni alloy
ZHANG Jian1, HUANG Ya-ni2, HUA Man-yu1, SHAO Yi-min1, ZHOU Dian-wu3, PENG Ping2
(1. Institute of Automobile and Mechanical Engineering,
Changsha University of Science and Technology, Changsha 410114, China;
2. College of Materials Science and Engineering, Hunan University, Changsha 410082, China;
3. State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body,
Hunan University, Changsha 410082, China)
Abstract:The influence and micromechanism of Al or Ti doping on the phase structural stability of Mg2Ni hydrogen storage alloy were investigated by the first-principles pseudopotential plane-wave method. The results show that within the range of doping concentration x from 0 to 0.5, the phase structural stability of Mg2Ni-type Mg2-xMxNi(M=Al, Ti) solid solution alloys is improved with increasing Al concentration. By contrast, the stability is weakened with increasing Ti concentration. These solid solution alloys all exhibit thermal instability with respect to the cubic Mg3MNi2(M=Al, Ti) compounds. They are likely to decompose into the multi-phases as composed of cubic Mg3MNi2(M=Al, Ti) and hexagonal Mg2Ni, which are in good agreement with the experimental results. The analysis of electronic structures shows that the phase structural stability of Mg2Ni hydrogen storage alloy with Al or Ti doping is closely associated with the bonding electron number of the doping systems, which originate mainly from lower energy ranges below Fermi energy level.
Key words: Mg2Ni; hydrogen storage alloy; phase structural stability; pseudopotential plane-wave; electronic structure
Mg2Ni合金因具有储氢容量高(3.6%,质量分数)、材料来源丰富、易合成及较纯Mg具有良好的吸/放氢动力学性能等优点,被认为是最具开发前途的金属储氢材料之一。然而,该体系较高的脱氢反应焓(64 kJ/mol)使其放氢温度仍然偏高,离实际应用尚存在一定距离[1-3]。为改善Mg2Ni合金的放氢性能,组元部分替代被证明是最有效的途径之一,如以Al、Ti和Nd等部分取代Mg2Ni中的Mg[4-6],或以Cu、Mn和Co等部分取代Mg2Ni中的Ni[7-9],均可明显提高体系的放氢能力。事实上,采用组元部分替代能够改善Mg2Ni体系放氢性能的主要原因在于合金化元素的掺杂,调整了Mg2Ni合金及其氢化物的相结构稳定性,使其脱氢反应焓减小,从而达到改善体系放氢性能的目的[10]。
近年来,人们针对Mg2Ni体系的相关性能也开展了一定的理论研究。如H?USSERMANN等[10]采用赝势平面波方法考察了Mg2NiH4氢化物的成键特性及其稳定性,发现Mg2NiH4中包含一种具有四面体结构且带有18个电子的NiH44-阴离子,且认为Al部分取代Mg可明显削弱氢化物的结构稳定性。JASEN等[11]采用密度泛函理论计算方法考察了Mg2NiH4氢化物的电子结构,发现Ni―H相互作用明显强于Mg―H的,且其主要表现为Ni sp与H s轨道电子的成键作用。马树元等[12]采用第一原理方法考察了Mg2Ni及其氢化物的电子结构,发现在Mg2Ni合金中,Ni―Ni间相互作用明显强于Mg―Ni的,且Mg2NiH4氢化物的稳定性与Mg2+与NiH44-的离子相互作用密切相关。ZENG等[13]采用从头算法考察了元素Al、Ag对Mg2NiH4氢化物脱氢反应焓的影响,发现元素Al或Ag部分取代氢化物中的Mg位,均降低了氢化物的稳定性,改善了体系的脱氢热力学行为。然而,以往的研究主要局限于Mg2NiH4氢化物相的成键特性及其放氢性能的 合金化效应,而关于Mg2Ni合金相结构及其稳定性方面的报道则较少。由上述分析得知,Mg2Ni合金本身的相结构稳定性也与该体系的放氢性能密切相关,为更好地理解合金化元素掺杂对Mg2Ni体系放氢性能的影响,本文作者采用第一性原理赝势平面波方法,选取Al或Ti作为掺杂元素,并基于已有的实验结果[4],将其部分取代Mg2Ni中的Mg,考察合金化元素对Mg2Ni合金相结构稳定性的影响,并从电子结构角度对其微观机理进行探讨,为改善Mg2Ni合金的放氢性能提供理论指导。
1 计算模型与方法
Mg2Ni的晶体结构呈密排六方Ca型结构,如图
1(a)所示,其单胞的最高对称性为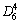 ,空间群为
,空间群为
P6222(No.180),晶格常数a=5.219 0 ?,c=13.293 0 ?[7]。Mg2Ni单胞是一种含有12个Mg原子和6个Ni原子的层状结构(6层),每隔一层含有2个Ni原子,分别占据晶格的3b(Ni1)和3d(Ni2)位;而每2个Ni原子层间含有4个Mg原子,分别占据晶格的6f (Mg1)和6i(Mg2)位。Mg2Ni晶胞中Mg与Ni的原子坐标分别为
+6Mg1:(0.5, 0, z),z=1/9;
+6Mg2: (x, 2x, 0),x=1/6;
+3Ni1:(0, 0, 1/6);
+3Ni2:(0, 0.5, 1/6)。
将2个Al或Ti分别取代Mg2Ni单胞中的2个Mg原子,得到Mg1.67Al(Ti)0.33Ni固溶体晶胞模型,如图1(b)所示;将3个Al或Ti分别取代Mg2Ni单胞中的3个Mg原子,得到Mg1.5Al(Ti)0.5Ni固溶体晶胞模型,如图1(c)所示;吕光烈等[4]通过实验研究发现,当Al或Ti掺杂于Mg2Ni中时,所形成的Mg2-xMxNi(M=Al,Ti)体系并非单一的六方晶型Mg2Ni结构,而是一种多相共存的合金,其中,存在一种具有立方结构的Mg3MNi2(M=Al,Ti)新型化合物,为此,本文作者也构建了该化合物的晶体结构[4],如图1(d)所示,其空间群为Fd3m(No.227),其中,Mg3AlNi2的晶格常数a=11.547 4 ?,Mg3TiNi2的晶格常数a=11.617 8 ?,Mg3MNi2(M=Al,Ti)晶胞中含有48个Mg原子,16个M(M=Al,Ti)原子和32个Ni原子,分别占据晶格的48f(Mg),16d(M)与32e(Ni)位,Mg、M(M=Al,Ti)及Ni在Mg3AlNi2与Mg3TiNi2晶胞中的原子坐标分别为
Mg3AlNi2:+48Mg (0.4298,0.1250,0.1250);
+16Al (0,0,0);
+32Ni (0.2058,0.2058,0.2058)。
Mg3TiNi2:+48Mg(0.4305,0.1250,0.1250);
+16Ti (0,0,0);
+32Ni (0.2067,0.2067,0.2067)。
在立方结构Mg3MNi2(M=Al,Ti)晶胞中,M(M=Al,Ti)原子所占比例正好与图1(c)所示的六方结构Mg1.5Al(Ti)0.5Ni固溶体晶胞中M(M=Al,Ti)原子掺杂浓度相同,M(M=Al,Ti)与Mg的摩尔比均为1:3。为便于计算,本模拟选取Mg3MNi2(M=Al,Ti)原胞作为计算模型,如图1(e)所示,可见,Mg3MNi2(M=Al,Ti)原胞中分别含有12个Mg原子,4个Al原子和8个Ni原子。
计算采用基于密度泛函理论的第一性原理赝势平面波方法[14],晶体波函数由平面波基组展开,交换关联能采用广义梯度近似GGA中的PBE关系式[15], 赝势为倒易空间表述的超软赝势[16], 动能截断点选取310.0 eV。总能计算采用自洽迭代方法,自洽计算时应用Pulay密度混合法[17],并应用基集修正[18] 。计算Mg、Ni、Al和Ti的赝波函数分别为Mg 2p63s2、Ni 3d84s2、Al 3s2 3p1和Ti 3d24s2。计算模型中,仅对Mg2Ni单胞(见图1(a))和Mg3MNi2(M=Al,Ti)原胞(见图1(e))进行完全弛豫;对于Mg1.67Al(Ti)0.33Ni(见图1(b))与Mg3Al(Ti)Ni2(见图1(c))固溶体晶胞,则仅对其胞内原子坐标进行弛豫,不弛豫其晶胞,固溶体晶胞的晶格常数均与Mg2Ni实验值[7]保持一致。体系弛豫时,总能量的收敛值取2.0×10-5eV/atom, 每个原子上的力小于0.8×10-10 N(0.05 eV/?), 公差偏移小于2.0×10-3 ?,应力偏差小于0.1 GPa;计算单点能时,体系总能量的收敛值取2.0×10-6 eV/atom。
2 计算结果与讨论
2.1 平衡晶格常数
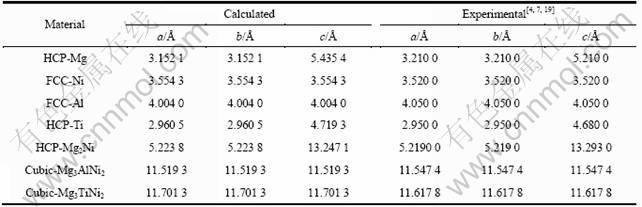
为验证计算方法与条件选取的合理性,首先测试研究体系所包含的固态金属(Mg,Ni,Al,Ti),六方结构Mg2Ni合金及立方结构Mg3MNi2(M=Al,Ti)化合物的平衡晶格常数,结果如表1所示。可见,计算所得固态金属的平衡晶格常数与各自的实验值[19]非常接近。同时,Mg2Ni和Mg3MNi2(M=Al,Ti)的计算结果与实验值[4, 7]也基本吻合,如Mg3AlNi2和Mg3TiNi2原胞完全优化后,转换成晶胞的平衡晶格常数分别为11.519 3 和11.701 3 ?,与其实验值 (11.547 4和11.617 8 ?) [4]十分接近,误差仅分别为0.24%与0.72%。由此看来,本文作者提出的计算方法与条件合理可靠。
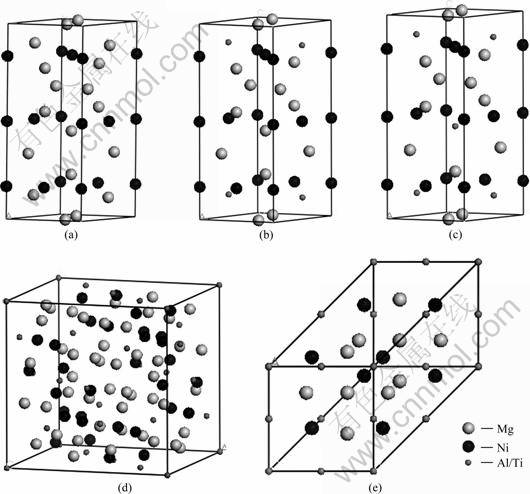
图1 Mg2Ni单胞、Mg1.67Al(Ti)0.33Ni晶胞、Mg1.5Al(Ti)0.5Ni晶胞、Mg3Al(Ti)Ni2晶胞和Mg3Al(Ti)Ni2原胞结构示意图
Fig.1 Schematic diagrams of structures of Mg2Ni unit cell (a), Mg1.67Al(Ti)0.33Ni crystal cell (b), Mg1.5Al(Ti)0.5Ni crystal cell (c), Mg3Al(Ti)Ni2 crystal cell (d) and Mg3Al(Ti)Ni2 primitive cell (e)
表1 固态金属Mg、Ni、Al、Ti和Mg2Ni合金及Mg3MNi2(M=Al,Ti)化合物的平衡晶格常数(a, b, c)
Table 1 Equilibrium lattice constants (a, b, c) of solid Mg, Ni, Al, Ti , Mg2Ni alloy and Mg3MNi2(M=Al,Ti) compounds
2.2 Al或Ti掺杂对Mg2Ni合金相结构稳定性的影响
合金形成热是衡量相结构稳定性的重要指标,合金形成热越负,对应合金的结构越稳定。为考察掺杂元素Al和Ti对Mg2Ni合金相结构稳定性的影响,本文作者首先计算Al或Ti掺杂于六方结构Mg2Ni合金前后及其具有立方结构Mg3MNi2(M=Al,Ti)化合物平均每个原子的合金形成热(ΔH),计算公式分别如式(1)和(2)所示[20]:
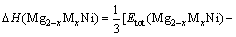
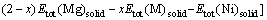 (1)
(1)
(M=Al, Ti;x=0, 0.33, 0.5)
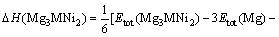
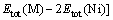 (2)
(2)
(M=Al, Ti;x=0, 0.33, 0.5)
式中:?H(Mg2-xMxNi)代表具有六方结构Mg2Ni(x=0)合金及Mg2-xMxNi (M=Al, Ti; x=0.33,0.5)固溶体合金的形成热;?H(Mg3MNi2)代表具有立方结构Mg3MNi2(M=Al, Ti)化合物的形成热;式(1)和(2)右边各项代表所对应的金属单质及合金每个化学式单元的总能量,其值与所有体系形成热的计算结果均列于表2。由表2可见,Mg2Ni合金形成热的计算值为-0.300 3 eV/atom, 而Al或Ti掺杂于Mg2Ni后,所形成六方结构固溶体合金的形成热相对于掺杂前发生了不同程度的变化。
表2 固态金属Mg、Ni、Al、Ti、Mg2-xMxNi(M=Al, Ti; x=0, 0.33, 0.5)固溶体合金及Mg3MNi2(M=Al,Ti)化合物的总能量(Etot)及合金形成热(ΔH)
Table 2 Total energies (Etot) and formation heat(ΔH) of solid Mg, Ni, Al, Ti, Mg2-xMxNi(M=Al, Ti; x=0, 0.33, 0.5) solid solution alloys and Mg3MNi2(M=Al, Ti) compounds

图2所示为 Al或Ti掺杂Mg2Ni合金的合金形成热。由图2可见,随着掺杂浓度的增加,所形成六方结构Mg2-xAlxNi(0<x≤0.5) 固溶体合金的形成热逐渐降低,而Mg2-xTixNi(02Ni体系的相结构稳定性,而Ti掺杂则相反,且随着掺杂浓度的增加,两者对Mg2Ni体系相结构稳定性增强或削弱的趋势逐渐明显。当Al或Ti的掺杂浓度x=0.5时,所形成六方结构Mg1.5Al(Ti)0.5Ni固溶体合金的形成热仍明显高于具有相同Al和Ti浓度立方结构Mg3Al(Ti)Ni2化合物的形成热。由于对于成分与浓度相同而结构不同的合金,合金形成热越负,对应的相结构越稳定[20],因此,当Al或Ti掺杂浓度x=0.5时,所形成的六方结构Mg1.5Al(Ti)0.5Ni固溶体合金更倾向于转变成立方结构的Mg3Al(Ti)Ni2合金而存在于自然界中。
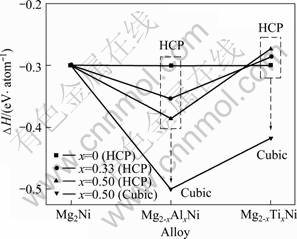
图2 Al或Ti掺杂Mg2Ni合金的合金形成热
Fig.2 Formation heat of Mg2Ni alloy doped with Al or Ti
2.3 Al或Ti掺杂Mg2Ni合金体系相对于Mg3MNi2(M=Al, Ti)化合物的稳定性
由上述计算得知,当Al或Ti掺杂浓度x=0.5时,所形成的六方结构Mg1.5Al(Ti)0.5Ni固溶体合金的稳定性低于具有立方结构Mg3Al(Ti)Ni2化合物的,在该浓度条件下合金相结构极易发生由六方结构向立方结构转变。那么,当Al或Ti掺杂浓度0<x<0.5时,所形成Mg2-xAl(Ti)xNi(0<x<0.5)固溶体合金相对于立方结构Mg3Al(Ti)Ni2化合物是否也呈现不稳定性,这是一个值得探讨的问题。为此,本文作者进一步研究了Mg2-xAl(Ti)xNi(0<x<0.5)固溶体合金相对于立方结构Mg3Al(Ti)Ni2化合物的热力学稳定性,可用如式(3)所示的化学反应来描述:

(M=Al, Ti; 0<x<0.5) (3)
由式(3)可知,若Mg2-xAl(Ti)xNi(0<x<0.5)固溶体合金相对于Mg3Al(Ti)Ni2化合物呈现热力学不稳定性,则式(3)化学反应为放热反应。为此,采用稳定性参量ΔE作为评估指标,公式如式(4)所示:
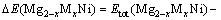
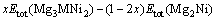 (4)
(4)
(M=Al, Ti; 0<x<0.5)
ΔE的计算结果见图3。由图3可见,当掺杂浓度0<x<0.5时,稳定性参量ΔE均大于0,表明所形成六方结构Mg2-xAl(Ti)xNi固溶体合金相对于立方结构Mg3Al(Ti)Ni2化合物均呈现热力学不稳定性,极易分解成由立方结构Mg3Al(Ti)Ni2与六方结构Mg2Ni组成的复合相。吕光烈等[4]研究表明,当Al或Ti部分取代Mg2Ni中的Mg后,所形成的Mg2-xAl(Ti)xNi合金已不是单一的六方晶型Mg2Ni结构,而是一种由立方结构Mg3Al(Ti)Ni2及六方结构Mg2Ni合金组成的多相结构,以上计算结果很好地解释了该实验现象。
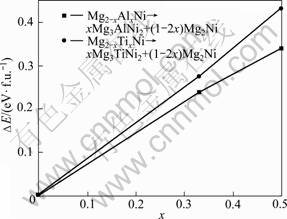
图3 Al或Ti掺杂Mg2Ni合金的热力学稳定性指标
Fig.3 Index of thermal stability of Mg2Ni alloy doped with Al or Ti
2.4 Al或Ti掺杂对Mg2Ni合金相结构稳定性的影响机理
为分析Al或Ti掺杂对Mg2Ni合金相结构稳定性的影响机理,以掺杂元素Al为代表,计算Al掺杂于Mg2Ni前、后及具有立方结构Mg3AlNi2化合物的总态密度及相应原子的分波态密度,图4(a)、(b)、(c)和(d)分别为具有六方结构的Mg2Ni、Mg1.67Al0.33Ni、 Mg1.5Al0.5Ni以及具有立方结构的Mg3AlNi2化合物平均每个原子的总态和分波态密度。可见,纯Mg2Ni体系的总态密度主要分布在-7.5~4 eV能量范围内(见图4(a)),其中,在-2.5~0 eV区间,成键电子主要来自Mg 3s,Mg 2p及Ni 3d的贡献;在-6~-2.5 eV区间,成键电子主要来自Mg 3s与Mg 2p的贡献;而在-7.5~-6 eV区间,成键电子则主要来自Mg 3s和少量Ni 4s的贡献。Al掺杂于Mg2Ni合金后,其态密度发生了明显变化(见图4(b)与(c)),主要表现如下:
1) Al掺杂使Mg2Ni体系的总态密度以及Mg和Ni原子的分波态密度均朝低能级方向偏移,如总态密度的分布范围已由掺杂前的-7.5~ 4 eV变为掺杂后的-8~ 4 eV,且随着Al元素掺杂浓度的增加,这种变化趋势则愈加明显;
2) 在费米能级以下的不同能量区间,成键电子除了来自Mg 3s、Mg 2p、Ni 4s和Ni 4d的贡献外,还有来自Al 3s与Al 3p电子的贡献。这样,一方面,成键电子数增多表明其价电子间相互作用增强,另一方面,更多的电子位于低能级区则使其体系结构变得更加稳定[21],因此,Al掺杂于Mg2Ni有利于提高体系的相结构稳定性。
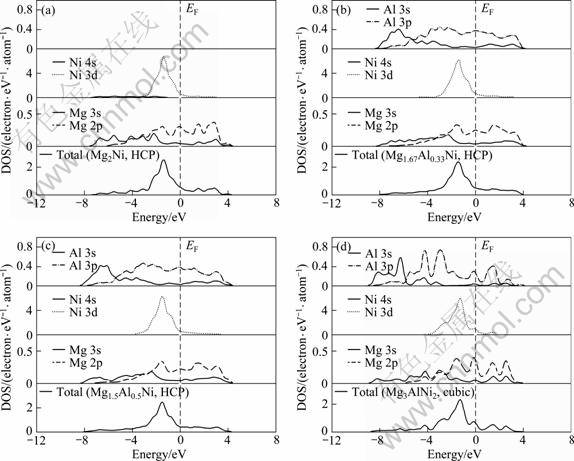
图4 Al掺杂前、后Mg2Ni体系及立方Mg3AlNi2化合物的总态及分波态密度
Fig.4 Total and partial densities of states of Mg2Ni systems without (a) and with Al doping (b), (c) as well as cubic Mg3AlNi2 compound (d)
进一步比较具有相同Al浓度的六方结构Mg1.5Al0.5Ni固溶体合金与立方结构Mg3AlNi2化合物的态密度(见图4(c)和(d))后发现,后者的总态密度以及Mg、Ni和Al原子的分波态密度进一步朝低能级区方向偏移,其总态密度的分布范围已拓宽为-9~4 eV,此外,在-2.5~0 eV,-3~-2.5 eV以及-5~-4 eV能量区间的成键峰高度相对于六方结构Mg1.5Al0.5Ni固溶体合金的均有增加,这可能是立方结构Mg3AlNi2化合物相对于六方结构Mg1.5Al0.5Ni固溶体合金以及其他低掺杂浓度Mg2-xAlxNi(0<x<0.5)固溶体合金具有更高相结构稳定性的根本原因[21]。
3 结论
1) 在掺杂浓度x=0~0.5范围内,所形成的Mg2Ni型Mg2-xMxNi(M=Al, Ti)固溶体合金的相结构稳定性 随Al掺杂浓度增大而增强,随Ti掺杂浓度增大而减弱。
2) Mg2-xMxNi(M=Al, Ti;x=0~0.5)固溶体合金相对于立方结构的Mg3MNi2(M=Al, Ti)化合物均呈现热力学不稳定性,极易分解成由立方结构Mg3MNi2(M=Al, Ti)和六方结构Mg2Ni组成的复合相。
3) 立方结构Mg3MNi2(M=Al, Ti) 化合物稳定性高于六方结构Mg2-xMxNi(M=Al, Ti;0<x≤0.5)固溶体合金的,其微观机理在于该化合物在低能级区具有更多的成键电子数。
REFERENCES
[1] BOUOUDINA M. Nanomaterials for hydrogen storage: Renewable and clean energy[J]. Int J Nanoelectronics and Materials, 2010, 3(2): 155-167.
[2] ZHAO X Y, MA L Q. Recent progress in hydrogen storage alloys for nickel/metal hydride secondary batteries[J]. Int J Hydrogen Energy, 2009, 34(11): 4788-4796.
[3] JURCZYK M, SMARDZ L, OKONSKA I, JANKOWSKA E, NOWAK M, SMARDZ K. Nanoscale Mg-based materials for hydrogen storage[J]. Int J Hydrogen Energy, 2008, 33(1): 374-38.
[4] 吕光烈, 陈林深, 胡秀荣, 王连邦, 袁华堂. Mg3MNi2(M=Ti, Al)的晶体结构[J].金属学报, 2001, 37(5): 459-462.
L? Guang-lie, CHEN Lin-shen, HU Xiu-rong, WANG Lian-bang, YUAN Hua-tang. The crystal structure of new hydrogen storage Mg3MNi2(M=Ti, Al) alloys[J]. Acta Metallurgica Sinica, 2001, 37(5): 459-462.
[5] ANIK M. Improvement of the electrochemical hydrogen storage performance of Mg2Ni by the partial replacements of Mg by Al, Ti and Zr[J]. J Alloys Compd, 2009, 486(1/2): 109-114.
[6] XIE D H, LI P, ZENG C X, SUN J W, QU X H. Effect of substitution of Nd for Mg on the hydrogen storage properties of Mg2Ni alloy[J]. J Alloys Compd, 2009, 478(1/2): 96-102.
[7] SIMI?I? M V, ZDUJI? M, DIMITRIJEVI? R, NIKOLI?-BUJANOVI? L, POPOVI? N H. Hydrogen absorption and electrochemical properties of Mg2Ni-type alloys synthesized by mechanical alloying[J]. J Power sources, 2006, 158(1): 730-734.
[8] ZHANG Y H, LI B W, REN H P, GUO S H, WU Z W, WANG X L. Hydriding and dehydriding characteristics of nanocrystalline and amorphous Mg20Ni10-xCox(x=0-4) alloys prepared by melt spinning[J]. Int J Hydrogen Energy, 2009, 34(6): 2684-2691.
[9] ZHANG Y H, LI B W, REN H P, GUO S H, ZHAO D L, WANG X L. Hydrogenation and dehydrogenation behaviours of nanocrystalline Mg20Ni10-xCux(x=0-4) alloys prepared by melt spinning [J]. Int J Hydrogen Energy, 2010, 35(5): 2040-2047.
[10] H?USSERMANN U, BLOMQVIST H, NOR?US D. Bonding and stability of the hydrogen storage material Mg2NiH4[J]. Inorg Chem, 2002, 41(14): 3684-3692.
[11] JASEN P V, GONZALEZ E A, BRIZUELA G, NAGEL O A, GONZALEZ G A, JUAN A. A theoretical study of the electronic structure and bonding of the monoclinic phase of Mg2NiH4[J]. Int J Hydrogen Energy, 2007, 32(18): 4943-4948.
[12] 马树元, 黄 丹, 郑定山, 肖荣军, 郭 进. Mg2Ni合金及其氢化物的电子结构及成键特性的第一原理计算[J]. 中国有色金属学报, 2007, 17(1): 118-123.
MA Shu-yuan, HUANG Dan, ZHENG Ding-shan, XIAO Rong-jun, GUO Jin. First principles calculation on Mg2Ni alloy and its hydride electronic structure and bonding characteristics[J]. The Chinese Journal of Nonferrous Metals, 2007, 17(1): 118-123.
[13] ZENG Y L, FAN K, LI X Y, XU B E, GAO X Z, MENG L P. First-principles studies of the structure and properties of Al- and Ag-substituted Mg2Ni alloys and their hydrides[J]. Int J Hydrogen Energy, 2010, 35(19): 10349-10358.
[14] LINDAN P L D, SEGALL M D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. J Phys: Condensed Matter, 2002, 14(11): 2717-2744.
[15] MARLO M, MILMAN V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals[J]. Phys Rev B, 2000, 62(4): 2899-2907.
[16] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41(11): 7892-7895.
[17] HAMMER B, HANSEN L B, NORKOV J K. Improved adsorption energetics within density-functional theory using revised perdew-burke-ernzerh of functionals[J]. Phys Rev B, 1999, 59(11): 7413-7421.
[18] FRANCIS G P, PAYNE M C. Finite basis set corrections to total energy pseudopotential calculations[J]. J Phys: Condensed Matter, 1990, 2(19): 4395-4404.
[19] KITTEL C. Introduction to solid state physics[M]. New York: Wiley, 1986: 30-80.
[20] ZHOU S H, NAPOLITANO R E. Phase stability for the Cu-Zr system: First-principle, experiments and solution-based modeling[J]. Acta Mater, 2010, 58(6): 2186-2196.
[21] ZHANG J, ZHOU D W, LIU J S. Study on hydrogen atom adsorption and diffusion properties on Mg(0001) surface[J]. Science in China (Series E): Technological Sciences, 2009, 52(7): 1897-1905.
(编辑 陈卫萍)
基金项目:长沙理工大学人才引进基金资助项目(20091026);长沙理工大学重点学科建设资助项目(08-007)
收稿日期:2010-05-13;修订日期:2010-12-10
通信作者:张 健,讲师,博士;电话:0731-85258630;E-mail:zj4343@126.com

