稀有金属 2008,(03),327-332 DOI:10.13373/j.cnki.cjrm.2008.03.001
双β-二酮桥联双核钯 (Ⅱ) 配合物的合成与谱学研究
黄可龙
中南大学化学化工学院,中南大学化学化工学院 湖南长沙410083 宜春学院江西省高校应用化学与化学生物学重点实验室,江西宜春336000,湖南长沙410083
摘 要:
以二氯化钯、联吡啶 (bpy) 、双乙酰丙酮合成了钯 (Ⅱ) 的新型固态三元桥联配合物[{Pd (C10H8N2) }2 (C10H12O4) ][NO3]2.2H2O。用元素分析、电导率、氢核磁共振、电喷雾质谱、红外光谱、电子光谱和荧光光谱对其进行了表征, 确定了配合物的组成。讨论了配合物在氮气气氛中的热分解行为。结果表明:该配合物属于低自旋的平面正方形, 配合物中离域π键和螯合环的形成使Pd-O和Pd-N键加强, 配合物很稳定。
关键词:
桥联双乙酰丙酮 ;双核钯配合物 ;合成 ;结构表征 ;热分析 ;
中图分类号: O641.4
收稿日期: 2007-04-28
基金: 江西省教育厅科技项目 (赣教技字[2006]338号); 宜春市高新技术产业重大项目 (JXYC2007KSA030) 资助;
Synthesis and Spectroscopy of Di-palladium (Ⅱ) Complex with Bis (β-diketonato) Bridged Ligand
Abstract:
A novel Bisacetylacetonate-bridged di-palladium (Ⅱ) complex defined by an O, O'-bidentate bisacetylacetonate dianion ligand and a chelating 2, 2'-bipyridine ligand, [{Pd (C10H8N2) }2 (C10H12O4) ][NO3]2.2H2O (C10H8N2=2, 2'-bipyridine; C10H12O4=Bisacetylacetonate dianion) , was synthesized and characterized by means of element analysis, conductivity, 1H NMR, ESI-MS, infrared spectra, electronic spectra and fluorimetry spectra. The results showed that the complex was a low-spin square planar coordinate compound. Thermogravimetry (TG) and differential scanning calorimetry (DSC) for the complex in an atmosphere of N2 were performed. In the complex the chelating ring, conjugated π bonds were formed, which further strengthened the Pd-O and Pd-N coordinate bonds and made Pd (Ⅱ) complex a stable compound.
Keyword:
bridged disacetylacetonate; dinuclear palladium (Ⅱ) complex; synthesis; characterization; thermal analysis;
Received: 2007-04-28
目前对钯 (Ⅱ) 配合物的研究除了作为均相催化反应和光催化反应的重要催化剂
[1 ,2 ]
以及抗肿瘤活性
[3 ,4 ]
研究之外, 主要集中在化学气相沉积技术 (CVD) 镀钯工艺
[5 ]
的研究。 随着CVD的日趋成熟, β-二羰基钯作为一种贵金属有机配合物, 因其可升华、 分解温度低、 易溶于多种有机溶剂等特点, 成为CVD 镀钯工艺的理想前驱体, 通过CVD可制备钯金属涂镀层、 载体催化剂、 膜催化剂以及钠米钯和钯合金。 如: Xomeritakis G等
[6 ]
以乙酰丙酮钯为原料, 用常压金属有机化合物化学气相沉积 (MOCVD) 过程在α-Al2 O3 管孔内制备钯膜获得成功, 自此开拓了其在新材料和精细化工领域的广泛应用和研究高潮
[7 ]
。 但对于钯 (Ⅱ) -双β-二酮类配合物, 尤其是双β-二酮桥联的双核钯 (Ⅱ) 类配合物研究很少
[8 ]
。
本文以联吡啶 (bpy) 为第一配体, 二乙酰丙酮为第二配体自组装合成了双核钯 (Ⅱ) 的新型固态三元配合物, 采用红外光谱 (IR) 、 电子光谱、 核磁共振光谱 (1 H NMR) 等手段, 表征了其化学结构及特征。 用TG和DSC研究了配合物在空气气氛下的热化学行为, 为CVD 镀钯工艺提供热学数据与参考。
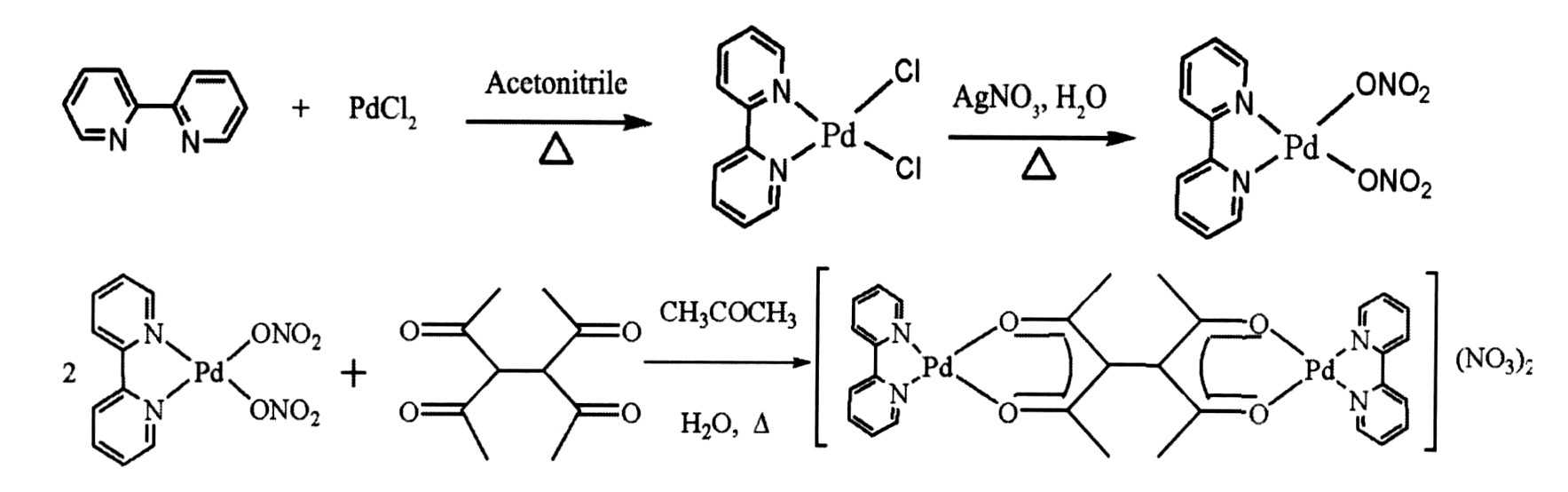
配合物的合成路线如下:
1 实 验
1.1 主要试剂与仪器
氯化钯 (沈阳市金科试剂厂, 分析纯, 钯含量≥59.0%) , 其他所使用的试剂均为分析纯。
元素分析: Flash EA1112 元素分析仪; 1 H NMR: Bruker Avance DMX400 (400 MHz) ; 紫外可见光谱: Shimadzu UV-2550型紫外可见分光光度计; 红外光谱: Bruker TENSOR 27型红外光谱仪 (KBr压片法, 4000~400 cm-1 ) ; 电喷雾质谱: Finnigan LCQ质谱仪和Thermo Finnigan DECAX-3000 LCQ Deca XP质谱仪 (乙腈-甲醇作流动相) ; 荧光光谱: PerkinElmer LS55型荧光分光光度计; 热分析: TA DSC-Q10型差示扫描量热分析仪和TA TGA-Q50型热重分析仪; 干燥器: DZF-2001型真空干燥器 (上海浦东荣丰科学仪器有限公司) ; 电导率仪: DDS-11A型数显电导率仪 (上海雷磁新泾仪器有限公司) ; 循环泵: DLSB-5L/40低温冷却循环泵 (巩义市予华仪器有限责任公司) 。
1.2 配体二乙酰丙酮的合成
参考文献
[
9 ]
的方法, 改成在水溶液中直接用NaOH代替乙醚溶剂中NaH来合成: 将40.0 g (1 mol) NaOH溶于50 ml去离子水中, 剧烈放热, 待其冷却后加入200 ml甲醇; 电磁搅拌下, 加入110 ml乙酰丙酮, 反应放热, 冷却后得到黄色晶体; 过滤, 固体用冰甲醇洗涤后再用甲醇重结晶, 得到白色晶体, 真空干燥, 得白色粉末状乙酰丙酮钠120.0 g。
称取24.4 g (0.2 mol) 乙酰丙酮钠悬浮于300 ml乙醚中, 25.4 g (0.2 mol) I2 溶于300 ml乙醚中, 然后滴加到乙酰丙酮钠悬浮液中, 电磁搅拌, 刚开始无变化, 1 h后变黄, 3 h后变为黄褐色, 停止搅拌, 底层为白色固体, 上层为黄褐色溶液; 过滤, 固体用500 ml去离子水洗, 干燥, 经甲醇重结晶, 得块状晶体, 置于真空烘箱60 ℃干燥12 h, 得二乙酰丙酮6.2 g, 产率为31%, M.P 191~192 ℃, 与文献值
[
9 ]
一致。 IR (KBr) : 1593 s (br) , 1413 s, 1364 (sh) , 1255 s, 1020 s, 1002 s。 1 H NMR (CD3 CN, 400 MHz) : 16.79 (2H, CH) ; 2.01 (s, 12H, CH3 ) 。 ESI-MS (m/z) : 199 (M+H+ , 100%) 。 λ max /nm (CH3 OH, 1.0×10-4 mol・L-1 ) : 286, 353。
1.3 配合物的合成
1.3.1 (2, 2′-联吡啶) 氯化钯的合成
1.0 g (5.64 mmol) 氯化钯溶于150 ml热乙腈中 (有部分黑色杂质, 过滤, 得清液) , 加入到250 ml单口瓶中。 电磁搅拌下, 880.9 mg (5.64 mmol) 2, 2′-联吡啶溶于少量乙腈中, 滴加到反应瓶中, 立即生成黄色针状晶体, 继续回流2 h。 然后冷却至室温, 过滤, 分别用少量乙腈和丙酮洗, 干燥, 得产物1.7 g, 产率88.3%。
1.3.2 (2, 2′-联吡啶) 硝酸钯的合成
1.7 g (5.0 mmol) (2, 2′-联吡啶) 氯化钯悬浮于200 ml pH=1的稀硝酸中, 电磁搅拌下滴加入1.7 g (5.0 mmol) AgNO3 的少量去离子水溶液, 于60 ℃避光反应8 h。 静置2 h, 过滤, 滤液旋蒸浓缩, 冷却结晶, 再过滤, 真空干燥, 得黄色固体1.74 g, 产率 90.0%。
1.3.3 (2, 2′-联吡啶) 硝酸钯与二乙酰丙酮的合成
77.3 mg (0.2 mmol) (2, 2′-联吡啶) 硝酸钯和19.8 mg (0.1 mmol) 二乙酰丙酮加入到50 ml单口瓶中, 再加入12 ml去离子水和6 ml丙酮; 电磁搅拌, 得到淡黄色澄清溶液, 室温下组装3 h。 然后在60 ℃下反应2 h, 冷却后过滤, 加入5 ml无水甲醇, 旋转蒸馏得到黄色固体, 少量冷水和乙醚洗涤, 真空烘箱中60 ℃干燥24 h, 得产物75.3 mg, 产率89.1%。 1 H NMR (CD3 OD, 400 MHz) : δ 2.26 (12H, s, CH3 ) , 7.87 (4H, t, J =8.5 Hz, bpy-H4, 4′ ) , 8.39 (4H, m, bpy-H3, 3′ ) , 8.52 (4H, m, bpy-H5, 5′ ) , 8.76 (4H, d, J =5.6 Hz, bpy-H6, 6′ ) 。 ESI-MS m /z : 779.8 (93%) , 872.8 (28%) , 262.8 (100%) 。 元素分析: 按C30 H32 N6 O12 Pd2 (FW: 881.46) 的实测值 (%) : C, 41.23; H, 3.79; N, 9.94。 计算值 (%) : C, 40.88; H, 3.66; N, 9.53。 摩尔电导Λ m (3.6×10-4 mol・L-1 乙腈溶液, 298 K) : 126.2 S・cm2 ・mol-1 。
2 结果与讨论
2.1 配合物的组成分析
从配合物的元素分析结果来看, 实验数据和理论值相吻合, 其摩尔电导数值也表明配合物为1∶2型电解质
[10 ]
。
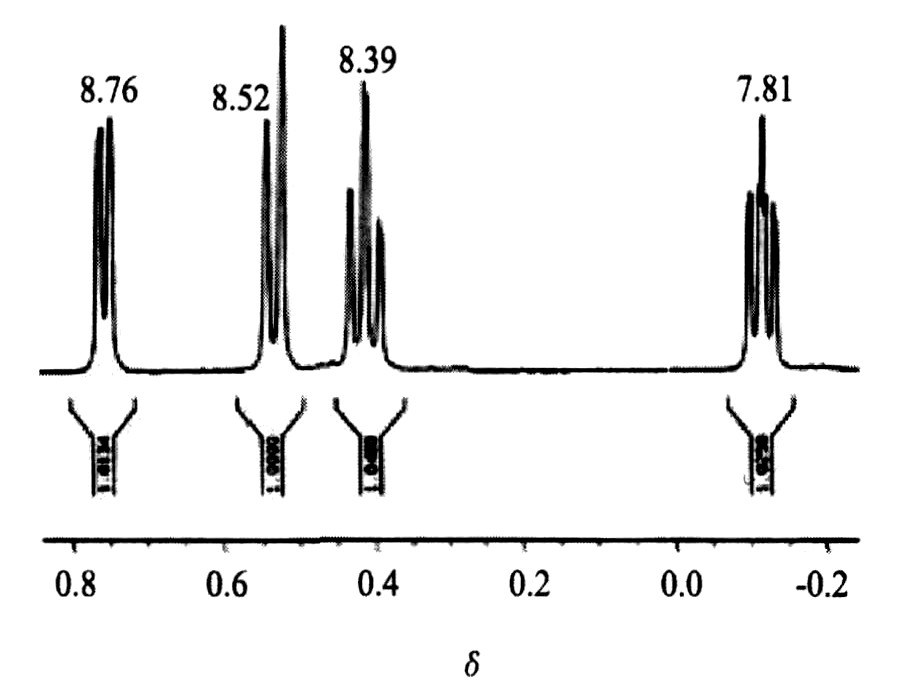
2.2 核磁共振氢谱分析
图1是配合物[{Pd (C10 H8 N2 ) }2 (C10 H12 O4 ) ][NO3 ]2 ・2H2 O的1 H NMR谱, 二乙酰丙酮与钯配位后形成了离域大π键, π键上的电子云向中心离子移动, 使氢核的电子云密度明显降低, 屏蔽效应减小, 化学位移增大, δ值增加, 并通过诱导效应影响到环外的甲基H。 另外, 在配合物分子中, 二乙酰丙酮根 (diacac2- ) 的4个氧分别与Pd2+ 配位而形成二个六元螯合环, 在这个环中, 由于离域π电子的存在而产生环电流效应。 螯合环与外磁场方向垂直时, 环电流产生的磁力线方向在环上、 下方与外磁场磁力线方向相反; 但在环侧面, 二者的方向则相同。 即环电流磁场增强了外磁场, 在环侧面的4个甲基氢氢核被去屏蔽, 共振谱峰位置移向低场, δ值由2.01增大到2.26 ppm。
在7.87, 8.39, 8.52, 8.76处质子峰分别对应于bpy中四组化学环境不等同的氢。
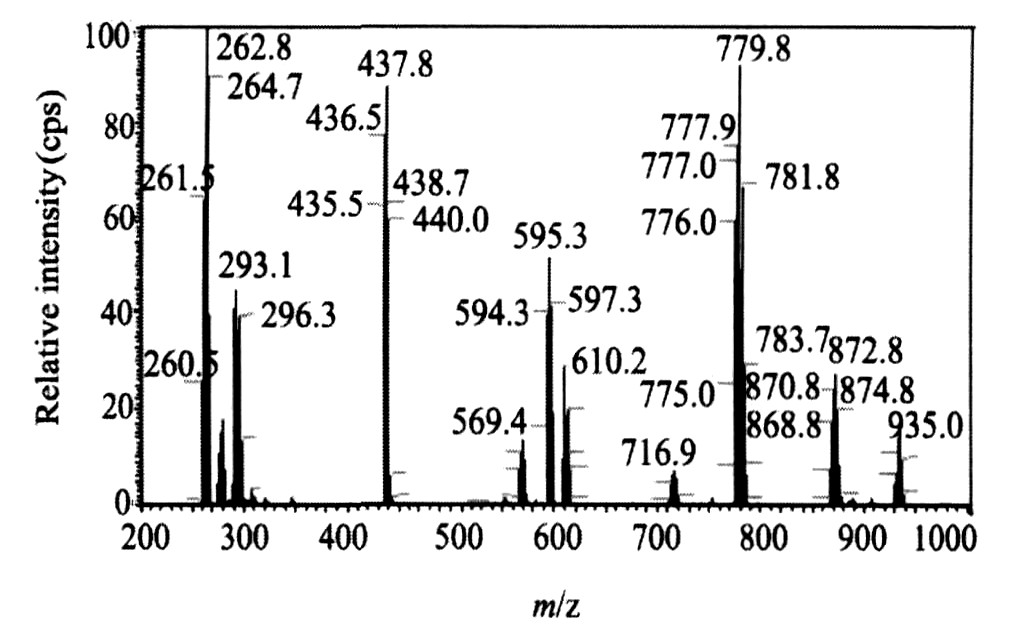
2.3 电喷雾质谱分析
配合物的阳离子电喷雾质谱见图2。 在868.8 (17%) , 870.8 (25%) , 872.8 (28%) , 874.7 (19%) 处产生一组同位素峰, 可以归属为[M-2H2 O+Na+ ]+ 。 在777.9 (75%) , 779.8 (93%) , 781.8 (68%) , 783.7 (30%) 处产生一组具有钯原子同位素特征的峰, 对应于二价质点[{Pd (C10 H8 N2 ) }2 (C10 H12 O4 ) (NO3 ) (H+ ) ]2+ , 是桥联双核钯 (Ⅱ) 配合物失去一个NO3 - 形成的。
2.4 红外光谱分析
配合物的红外光谱的观测频率和谱带归属见表1。 由这些谱带归属和比较可知, 在Pd样品分子中不仅存在着OH, bpy, CH3 , C=C, C=O, C-CH3 , NO3 - 等基团, 同时也得到了Pd-N, Pd-O键及螯合环的红外特征光谱。
配合物在466, 556 cm-1 处出现ν Pd-N 吸收峰, 可归属为Pd-N伸缩振动
[11 ]
; 配合物在1384 cm-1 出现了自由的NO3 -1 的特征吸收峰。 比较该配合物与bpy的红外光谱发现, bpy形成配合物后其自由配体位于3055 cm-1 处的ν C-H 振动吸收峰消失, 其骨架振动峰ν C=C 和ν C=N 由1454和1418 cm-1 移至1476和1465 cm-1 , 且1476 cm-1 处振动峰减弱, bpy环上相邻H原子的同位相面外弯曲振动δ C-H 由759移至770 cm-1 。 吸收峰蓝移, 是由于bpy的两个氮原子与Pd2+ 配位后, 吡啶环的呼吸振动受阻、 能量升高所致
[12 ]
。
图1 配合物[{Pd (C10H8N2) }2 (C10H12O4) ][NO3]2・2H2O的1H NMR谱 (横坐标加位移符号) Fig.1 1HNMR spectra of the complex[{Pd (C10H8N2) }2 (C10H12O4) ][NO3]2・2H2O
图2 配合物[{Pd (C10H8N2) }2 (C10H12O4) ][NO3]2・2H2O的阳离子ESI-MS谱Fig.2 ESI-MS spectra of the complex[{Pd (C10H8N2) }2 (C10H12O4) ][NO3]2・2H2O
表1 配合物的红外光谱频率和谱带归属
Table 1 IR absorptance data of the title complex
Wave number/cm-1
Attribution
Wave number/cm-1
Attribution
ν (OH) 1020
ρ (CH3 )
ν (CH3 ) 943
ν (C=C) +ν (C=O)
ν (C=O) +ν (C=O) 770
π (CH)
ν (C=C) +ν (C=N) 718
ν (C-CH3 +cyclic distortion
δ (CH3 ) 665
(Pd-O) +π (H3 C-CCO)
NO3 -
556
ν (Pd-N)
ν (C-CH3 ) +ν (C=C) 466
ν (Pd-N) +ν (Pd-O) +ν (C-CH3 )
δ (C-H) +ν (C-CH3 ) 418
Cyclic distortion+ν (Pd-O)
Diacac2- 与二个Pd2+ 配位后, 分别形成了大π键, β-二酮单元的两个羰基形成六元鳌环, 环上电子云往中心离子的方向移动, 使得C=O键减弱, 已不是典型的羰基, 观察不到明显的典型羰基的伸缩振动吸收, 而烯醇式羰基的伸缩振动峰一般在1600 cm-1 附近, 本配合物中该吸收峰和bpy环的骨架伸缩振动吸收峰相重叠, 烯醇羰基的伸缩振动分裂为1601和1551 cm-1 处, 说明该配合物中Diacac2- 以烯醇式二价阴离子形式与Pd2+ 离子发生配位, 1601和1551 cm-1 处分别为Diacac2- 上C=O和C=C键的伸缩振动峰; 在3100~2850 cm-1 范围内的宽吸收峰, 主要是由该混配物中Diacac2- 上甲基、 烯键=C-H和bpy的芳环上C-H的伸缩振动所致, 在3022和2926 cm-1 处的峰型较明显; 1415和1336 cm-1 处的两个吸收峰分别是Diacac2- 上甲基的反对称变形振动和对称变形振动。 1265 cm-1 处的吸收峰为C-CH3 的伸缩振动和C=C键的伸缩振动的偶合所致。 1020 cm-1 的吸收峰可归属于Diacac2- 上甲基的C-H的摇摆振动峰。 另外, 在IR谱中还出现665, 466, 418 cm-1 等峰, 这些峰可以归属为Pd-O键的伸缩振动以及螯合环的变形振动, 表明Diacac2- 中的O与Pd2+ 形成了配位键并已成环
[13 ]
。 3446 cm-1 处的强宽峰表明配合物中存在水分子, 热分析数据也证实配合物中存在二个水分子。
2.5 电子光谱分析
二乙酰丙酮类过渡金属配位化合物分子中可能存在L→M电荷跃迁、 M→L电荷跃迁、 配体的π→π* 跃迁和中心离子的d→d电子跃迁。 表2为配合物在CH3 CH2 OH溶剂中的紫外-可见吸收光谱。
由表2可知, 该配合物在紫外-可见光区出现了3个强吸收峰与1个肩峰。 Pd与配体发生了配位反应, 电子跃迁发生改变, 配体的π→π* 跃迁向短波方向迁移至235 nm处, 并由于其强度不及中心离子电荷跃迁而被掩盖成为肩峰。 配合物中心离子Pd2+ 为d8 电子构型, 与强场配体Diacac2- 以及bpy相结合时, 进行dsp2 杂化, 形成近似平面正方形配合物, 具有D4h 的对称性, 其中8个d电子占据能量较低的dxy yz xz z 2 四个轨道, 能级较高的dx 2 -y 2 轨道则空着。 在平面正方形场中存在种类型的d→d电子跃迁1 A1 →1 E (dxy x 2 -y 2 ) 和1 A2 →1 E (如dz 2 →dx 2 -y 2 ) , 由于两种跃迁所需能量比较接近, 强度也较小, 其紫外-可见吸收谱峰经常重叠在一起, 出现较宽的吸收带, 在较长的敞长310 nm处出现的宽峰应归属为两种d→d跃迁的叠加。 另外, 配合物在223和208 nm处出现的两个强吸收应分别归属为L→M跃迁和M→L跃迁, 因为这两种跃迁所需能量最大, 且不受自旋禁阻的影响, 多发生在波长短的地方, 强度大
[14 ]
。
表2配合物在CH3CH2OH溶剂中的紫外-可见吸收光谱 (浓度为9.94×10-5mol・L-1)
Table 2 UV-Vis absorptance spectra for CH 3 CH 2 OH solution of complex (concentration 9.94×10 -5 mol・L -1 )
λ max /nm (ε /L・mol-1 ・cm-1 ) Transition attribution
4 ) d→d
π→π*
4 ) L→M
4 ) M→L
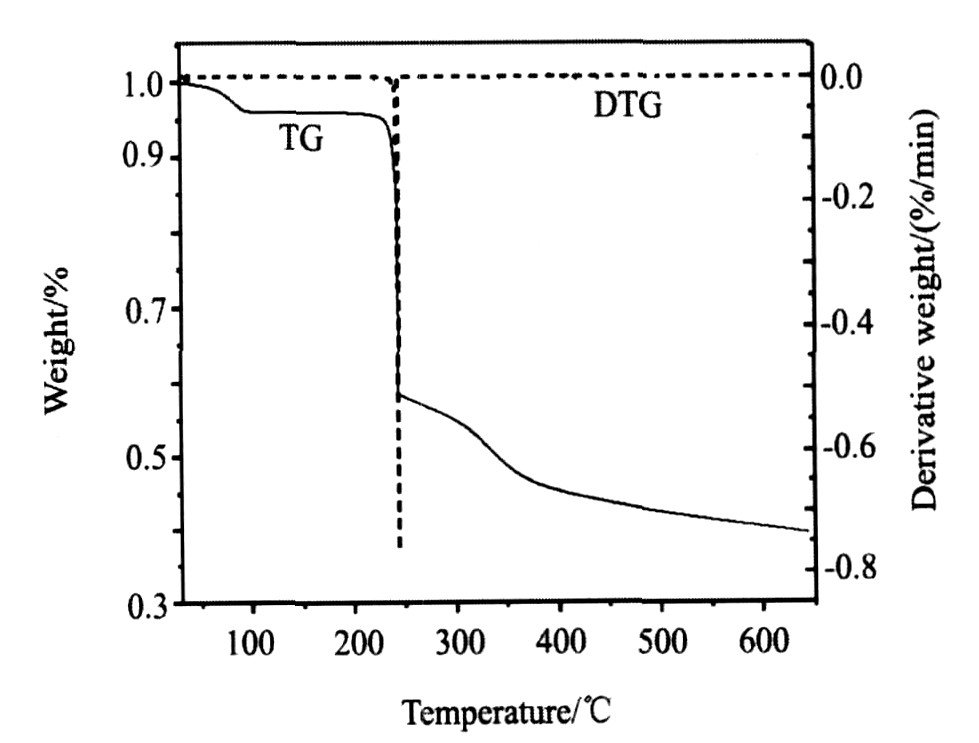
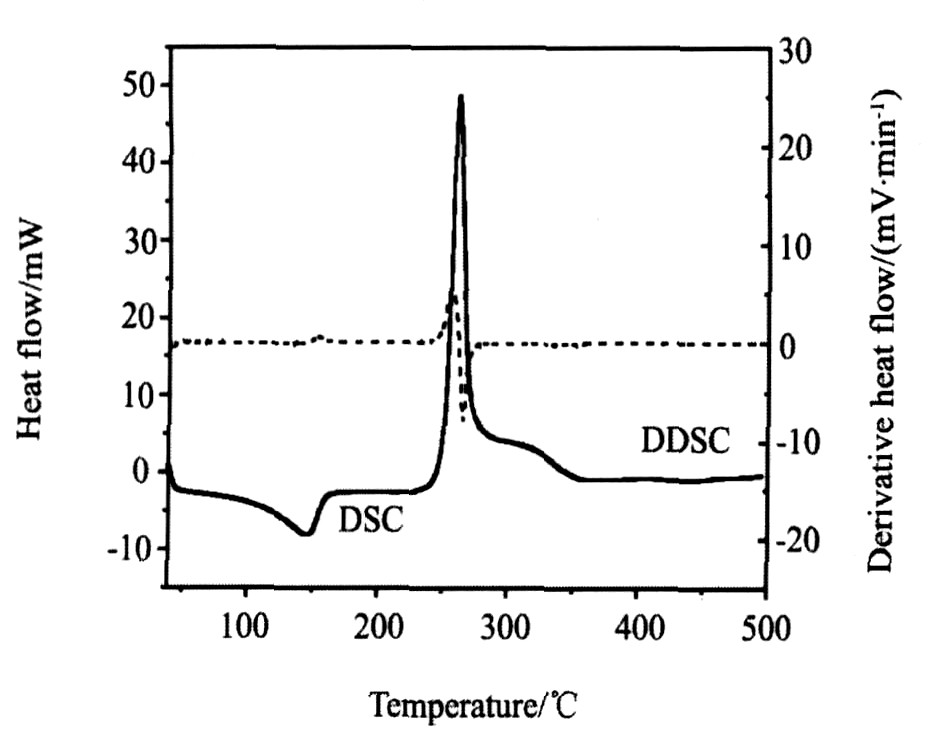
2.6 热谱分析
从图3配合物在氮气气氛中的TG-DTG曲线可知, 在30~650 ℃区间配合物有3个失重台阶, 第一步在41~101 ℃区间失去二个结晶水, 失重率为3.98% (理论值为4.09%) ; 第二步失去的是桥联配体二乙酰丙酮阴离子和二个NO3 - , 此阶段对应的分解温度及失重率分别为203 ℃和36.51% (理论值为36.34%) , 在243 ℃时TG曲线急剧下降, 开始急速失重, 同时在该温度点DTG曲线出现尖锐的放热峰, 并大量放热; 第三步随着温度的升高失重较慢, 失去的是一个端基配体2, 2′-联吡啶分子, 此阶段对应的分解温度及失重率分别为251 ℃和16.68% (理论值为17.73%) , 到544 ℃时TG及DTG曲线都趋于平稳, 产物为钯和一个2, 2′-联吡啶分子, 产物残余量为41.34% (理论值为41.88%) 。 实验值与理论值吻合较好, 进一步论证了该结构式是正确的。
图3 配合物的TGD-TG曲线
Fig.3 TG-DTG curve of title complex
DSC曲线表明 (图4) , 在不到146 ℃出现一个吸热过程 (Endothermic) , 随后在261 ℃出现一个放热过程 (Exothermic) 。 DDSC曲线说明DSC曲线的两个峰都是单独、 无重复的峰。
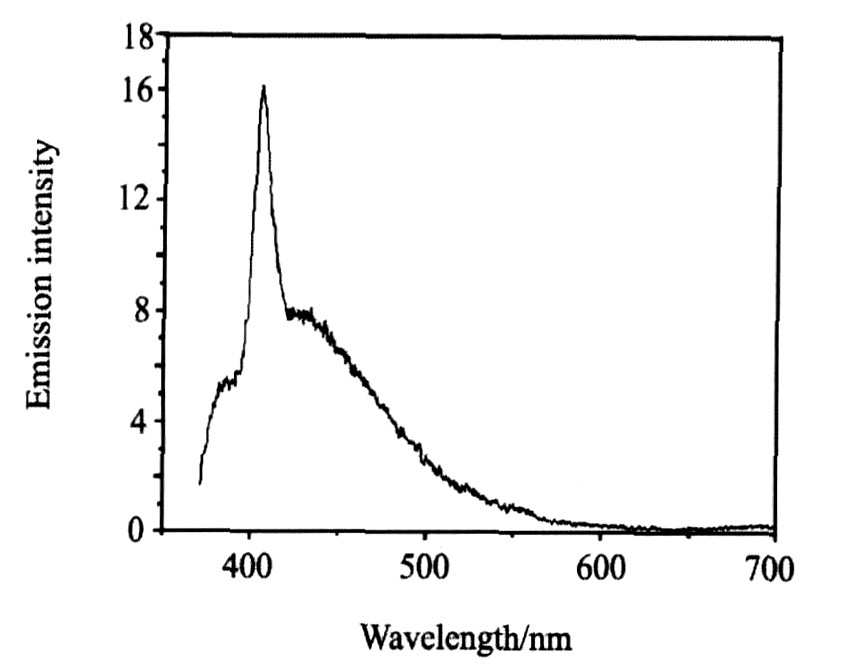
2.7 荧光光谱分析
配合物的荧光光谱如图5所示, 是在2.2×10-5 mol・dm-3 的甲醇溶液中氮气保护下用362 nm光激发室温测定的, 最大荧光发射波长为405 nm, 由1 MLCT[d (Pd) →π* (bpy) ]跃迁产生。
图4 配合物的DSC-DDSC曲线
Fig.4 DSC-DDSC curve of title complex
图5 配合物的荧光光谱 (2.2×10-5 mol・L-3的甲醇溶液)
Fig.5 Fluorimetry spectra of title complex (2.2×10-5 mol・L-1 in CH3 OH)
3 结 论
以联吡啶为第一配体, 二乙酰丙酮为第二配体, 自组装合成了双核钯 (Ⅱ) 的新型固态三元配合物, 自组装反应步骤的产率达到89.1%。 通过元素分析、 电导、 红外光谱、 电子光谱、 氢核磁共振光谱、 电喷雾质谱和荧光光谱等手段, 确证了所得产物的结构, 利用TG-DTG研究了配合物在氮气气氛下的热分解行为。 样品为平面正方形配合物, 两种含氮和氧的配体与Pd形成螯合环, 螯环内又构成离域大π键, 增加了配合物的稳定性。
参考文献
[1] Viciu MS, Kissling R M, Stevens E D, Nolan S P.An air-stable palladium/N-heterocyclic carbene complex and its reactivity in aryl amination[J].Organic Letters, 2002, 4 (13) :2229.
[2] Adjabeng G, BrenstrumT, Frampton CS, Robertson AJ, Hillhouse J, McNultyJ, Capretta A.Palladiumcomplexes of1, 3, 5, 7-tetra-methyl-2, 4, 8-trioxa-6-phenyl-6-phosphaadamantane:synthesis, crystal structure and use inthe suzuki and sonogashira reactions and the r-arylation of ketones[J].J.Org.Chem., 2004, 69 (15) :5082.
[3] Padhye S, Afrasiabi Z, Sinn E, Fok J, Mehta K, Rath N.Anti-tumor metallothiosemicarbazonates:Structure and antitumor activity of palladium complex of phenanthrenequinone thiosemicarbazone[J].Inorg.Chem., 2005, 44 (5) :1154.
[4] 袁晓玲, 曾锦萍, 梅光泉.贵金属配合物在医药领域中的应用[J].稀有金属, 2005;29 (4) :418.
[5] Frederick D Lewis, Gwen D Salvi.Platinum (Ⅱ) bis (β-diketo-nates) as photoactivated hydrosilation catalysts[J].Inorg.Chem., 1995, 34 (12) :3182.
[6] Xomeritakis G, Lin Y S.Fabrication of a thin palladium mem-brane supposedin a porous ceramic substrate by chemical vapor dep-osition[J].Journal of Membrane Science, 1996, l20 (2) :26l.
[7] Huang HP, Li S H, Yu S Y, Li YZ, Jiao Q, Pan YJ.Novel pyrazolate-bridged dinuclear Pd (Ⅱ) diimine complexesthat bindin-organic anions[J].Inorg.Chem.Commun., 2005, 8:656.
[8] Mei G Q, Huang K L, Huang HP.3, 3′-Bisacetylacetonato-bis[ (1, 10-phenanthroline) palladium (Ⅱ) ]bis (hexafluorophosphate) acetonitrile solvate[J].Acta Crystallographica Section E, 2006;E (62) :m2743.
[9] Kruger P E, Moubaraki B, Fallon GD, Murray KS.Tetranuclear copper (Ⅱ) complexes incorporating short andlong metal-metal sep-arations:synthesis, structure and magnetism[J].J.Chem.Soc, Dalton Trans., 2000, (5) :713.
[10] Geary WJ.Use of conductivity mesurementsin organic solventsfor the characterization of coordination compounds[J].Coord.Chem.Rev., 1971, 7 (1) :81.
[11] Newkome G R, Puckett WE, Kiefer GE, Gupta VK, Fronczek F R, Pantaleo D C, McClure G L, Simpson J B, Deutsch W A.Chemistry of heterocyclic compounds series.94.Square-planar cis-andtrans-C-palladium (Ⅱ) complexes of N electron-deficient het-eroaromatic ligands.Ligand synthesis, complexation, and spectralanalyses and complexinteraction with phage PM2DNA[J].Inorg.Chem., 1985, 24 (6) :8l1.
[12] 赵永亮, 赵风英.铕镧苯甲酸α, α′-联吡啶配合物的合成及性质研究[J].光谱学与光谱分析, 2002, 22 (6) :987.
[13] 中本一雄.无机和配位化合物的红外和拉曼光谱 (第四版) [M].北京:化学工业出版社, 1991.288.
[14] 梁广, 刘伟平, 普绍平, 高文桂, 闫革新.乙酰丙酮钯 (Ⅱ) 的合成及其结构表征[J].贵金属, 2004, 25 (4) :12.