Ce和V双掺杂对γ-TiAl基合金塑性和电子结构的影响
来源期刊:稀有金属2019年第9期
论文作者:宋庆功 朱燕霞 王丽杰 顾威风 胡雪兰 康建海
文章页码:942 - 951
关键词:γ-TiAl基合金;塑性;电子结构;化学键;第一性原理;
摘 要:γ-TiAl基合金是具有优异综合性能和重要应用价值的高温结构材料。室温塑性不足抑制了这类合金的广泛应用。原子替位双掺杂是改善其室温塑性的有效方法之一。采用基于密度泛函理论的第一性原理方法,计算研究了Ce和V原子替位双掺杂γ-TiAl基合金的几何结构、总能量、弹性常数、电子态密度、重叠布居数、电荷密度和差分电荷密度等,并计算了原子平均形成能、弹性模量比、赝能隙。由原子平均形成能和Born-Huang判据可知,各个Ce和V替位双掺杂γ-TiAl基合金体系都具有能量稳定性和力学稳定性。由弹性模量比预测,各个Ce和V替位双掺杂γ-TiAl基合金体系的塑性均有明显改善。通过对典型合金体系Ti8Al6CeV的重叠布居数和电荷密度分析,揭示材料塑性改善的内在因素是:Ce和V替位双掺杂使Al-3p与Ti-3d轨道间的共价结合显著弱化; Ti-d与Ti-d轨道间的杂化显著增强;而Al与Al原子间的共价结合明显弱化。
网络首发时间: 2018-12-20 07:07
稀有金属 2019,43(09),942-951 DOI:10.13373/j.cnki.cjrm.xy18080016
宋庆功 朱燕霞 王丽杰 顾威风 胡雪兰 康建海
中国民航大学理学院低维材料与技术研究所
中国民航大学中欧航空工程师学院
γ-TiAl基合金是具有优异综合性能和重要应用价值的高温结构材料。室温塑性不足抑制了这类合金的广泛应用。原子替位双掺杂是改善其室温塑性的有效方法之一。采用基于密度泛函理论的第一性原理方法,计算研究了Ce和V原子替位双掺杂γ-TiAl基合金的几何结构、总能量、弹性常数、电子态密度、重叠布居数、电荷密度和差分电荷密度等,并计算了原子平均形成能、弹性模量比、赝能隙。由原子平均形成能和Born-Huang判据可知,各个Ce和V替位双掺杂γ-TiAl基合金体系都具有能量稳定性和力学稳定性。由弹性模量比预测,各个Ce和V替位双掺杂γ-TiAl基合金体系的塑性均有明显改善。通过对典型合金体系Ti8Al6CeV的重叠布居数和电荷密度分析,揭示材料塑性改善的内在因素是:Ce和V替位双掺杂使Al-3p与Ti-3d轨道间的共价结合显著弱化; Ti-d与Ti-d轨道间的杂化显著增强;而Al与Al原子间的共价结合明显弱化。
中图分类号: TG146.23
作者简介:宋庆功(1958-),男,河北唐山人,博士,教授,研究方向:航空材料、金属间化合物;电话:022-24092516;E-mail:qgsong@cauc.edu.cn;
收稿日期:2018-08-11
基金:国家自然科学基金项目(51201181);中国民航大学自然科学基金项目(08CAUC-S02)资助;
Song Qinggong Zhu Yanxia Wang Lijie Gu Weifeng Hu Xuelan Kang Jianhai
Institute of Low Dimensional Materials and Technology,College of Science,Civil Aviation University of China
Sino-European Institute of Aviation Engineering,Civil Aviation University of China
Abstract:
γ-TiAl based alloys are high-temperature structural materials with excellent comprehensive properties and important application values, while their insufficient plasticity at room temperature inhibits the widespread use of such alloys. Atomic substitution co-doping is one of the effective methods to improve the plasticity of room temperature. The geometric structures, total energies, elastic constants, electronic densities of states, overlap populations, charge densities and charge density differences of Ce and V atoms substitution co-doping γ-TiAl based alloys were investigated by using the first-principles method based on the density functional theory. On this basis, the average formation energies of the atoms, elastic modulus ratios, and pseudo-gaps of the systems were calculated and studied. According to the average formation energy and Born-Huang criterion, all of the Ce and V substitution double doping γ-TiAl based alloys had energy stability and mechanical stability. The significant improvement of plasticity of Ce and V substitution co-doping γ-TiAl systems was predicted by their elastic modulus ratios. The analysis about the overlap population and charge density of the typical alloy system Ti8Al6CeV suggested the intrinsic factors that improved the plasticity of the materials were: the Ce and V atoms substitution co-doping significantly weakened the covalent bonds of Al-3 p and Ti-3 d orbitals. The hybridigation bonds between Ti-d and Ti-d orbitals were significantly enhanced, and the covalent bonds between Al and Al atoms were significantly weakened.
Keyword:
γ-TiAl based alloy; plasticity; electronic structure; chemical bond; first-principles;
Received: 2018-08-11
钛铝基合金具有比强度高、 熔点高、 密度低、 耐腐蚀性能好的特性, 且在高温条件下具有良好的抗氧化性和抗蠕变性能, 是业界一直期待的新一代高温结构材料之一
王海燕等
近年来, 通过掺杂稀土元素合金化来改善金属间化合物室温塑性的方法成为人们关注的焦点
1 结构模型和计算方案
1.1 结构模型
γ-TiAl属于金属间化合物, 晶体结构为L10型面心立方结构, 如图1(a)所示, 其晶胞内包含2个Ti原子和Al原子, 晶格参量为a0=b0=0.398 nm, c0=0.404 nm, α=β=γ=90°。 该结构可视为由一个纯Ti和一个纯Al的简单四方晶格相互套构而形成的复式格子, 如图1(b)所示, 相应的晶格参量为a=b=0.284 nm, c=0.406 nm。 为了便于计算研究, 以图1(b)为基础构建2×2×2超胞结构, 即Ti8Al8体系, 记为S0, 如图1(c)所示。
以S0为基础, 分别构造Ce替代Al且V替代Ti双掺杂体系S1 (Ti7VAl7Ce); Ce替代Ti且V替代Al双掺杂体系S2 (Ti7CeAl7V); Ce和V同时替代Al双掺杂体系S31~S35(Ti8Al6CeV);Ce和V同时替代Ti双掺杂体系S41~S45(Ti6CeVAl8),共12个双掺杂γ-TiA l体系。图2(a~f)为双掺杂体系的典型构型。
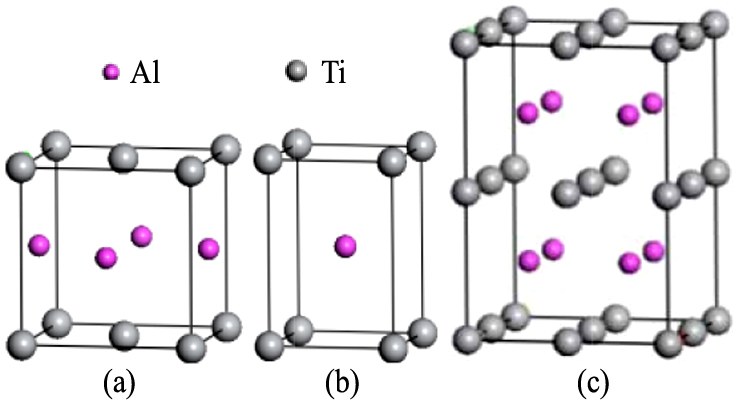
图1 γ-TiAl结构模型
Fig.1 Structural models of γ-TiAl
(a)Unit cell of L10structure;(b)Unit cell of simple tetragonal structure;(c)2×2×2 supercell of simple tetragonal structure
1.2 计算方案设置
采用基于密度泛函理论的第一性原理方法进行研究, 计算软件为Materials Studio 6.0的CASTEP (Cambridge sequential total energy package)模块, 利用高性能计算机集群进行相关计算和分析。 交换关联能选用广义梯度近似GGA (generalized gradient approximation)下的PBE (perdew-burke-ernzerhof) 泛函, 赝势取超软赝势来描述离子实与价电子之间的相互作用, 将Ti原子的3s23p63d24s2和Al原子的3s23p1视为价电子。 相应的计算参数设置如下: 晶体中电子波函数由平面波基组展开,平面波数目由动能截断值来决定,超胞所选取的动能截断值为350 eV,k点网格数取4×4×3。采用拟牛顿法对模型进行了结构的优化,来求得体系的局域最稳定结构。迭代计算收敛标准为:原子作用力低于0.3 eV・atom-1,原子能量变化值低于1.0×10-5eV・atom-1,原子的最大位移为0.0001 nm,应力偏差小于0.05 GPa。对所有掺杂体系进行几何优化获得稳定结果后,计算体系的能量、弹性常数、电子态密度及电荷密度等,并依据物理与化学理论进行分析讨论。
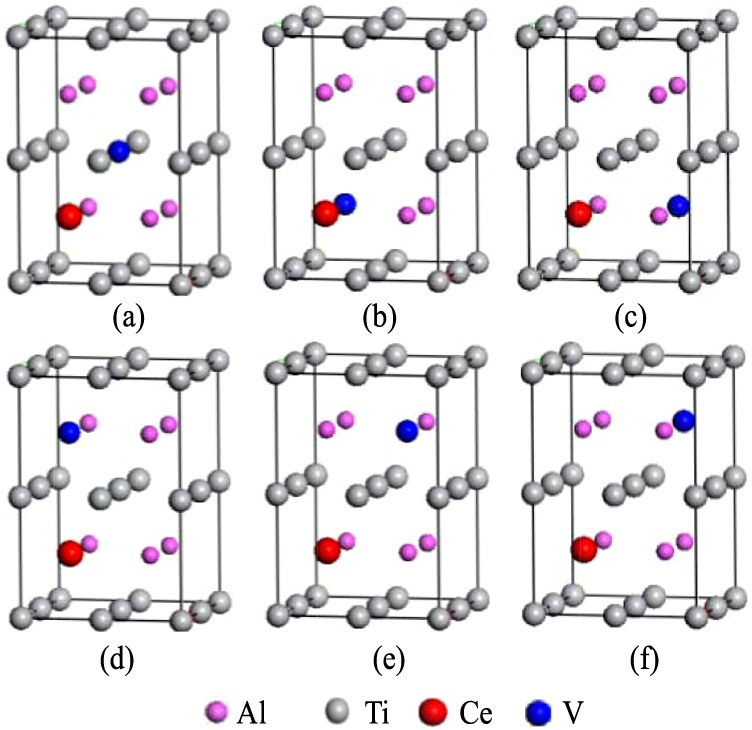
图2 Ce和V双掺杂γ-TiAl体系的典型构型
Fig.2 Typical structural models of Ce and V co-doping γ-TiAl systems
(a)S1;(b)S31;(c)S32;(d)S33;(e)S34;(f)S35
2 计算结果与分析
2.1 晶格结构及参量
经过几何优化后, 纯γ-TiAl体系S0的晶格参量为a=b=0.5638 nm, c=0.8208 nm, 与报道的实验值(2a0=0.5674 nm, 2c0=0.8118 nm)
由表1可以看出, 所有双掺杂体系的晶格参量a较纯γ-TiAl体系均有所增加; 但对于不同的双掺杂体系, 晶格参量c增减不一。 这将会导致双掺杂体系的轴比R(c0/a0)发生变化, 从而改变了合金体系的立方度。 这也是改善合金体系的室温塑性的重要方向。 易见, Ce和V替位双掺杂体系的空间群也分别变为正交和单斜体系, 对称性降低。 双掺杂体系的晶胞体积有所增大, 这主要是原子半径较大的Ce原子的影响, 这导致晶格发生畸变、 体积膨胀。 因此应尽量降低其在合金中的含量。 尽管如此, Ce和V双掺杂γ-TiAl体系的密度仍在4.5 g・cm-3以下。 这远小于镍基合金的密度(7.9~8.5 g・cm-3), 因而Ce和V双掺杂γ-TiAl体系在密度上仍有明显优势。 由于晶胞体积增大必将影响原子间化学键的强度, 在一定程度上会降低TiAl金属间化合物的塑性变形效果, 而且掺杂体系的对称性也降低。
2.2 形成能和稳定性
材料的稳定性可用平均形成能描述。 对于Ce和V双掺杂体系, 其原子平均形成能
Eb=(Et-mETi-nEAl-kECe-lEV)/N (1)
表1 Ce和V双掺杂γ-TiAl基合金体系的几何性质
Table 1 Geometrical properties of Ce and V co-doping γ-TiAl based alloy systems
| Systems | a/nm | b/nm | c/nm | Space group | Crystal system | Volume/nm3 | Density/(g・cm-3) |
S0 |
0.5638 | 0.5638 | 0.8208 | P4/mmm | Tetragonal | 0.260 | 3.8129 |
S1 |
0.5864 | 0.5864 | 0.7915 | Cm | Monoclinic | 0.273 | 4.3538 |
S2 |
0.5811 | 0.5811 | 0.8080 | Cm | Monoclinic | 0.271 | 4.3790 |
S31 |
0.6020 | 0.5769 | 0.7823 | Pm | Monoclinic | 0.272 | 4.5000 |
S32 |
0.5857 | 0.5857 | 0.7916 | Amm2 | Orthorhombic | 0.272 | 4.5021 |
S33 |
0.5899 | 0.5899 | 0.7841 | Amm2 | Orthorhombic | 0.273 | 4.4808 |
S34 |
0.5914 | 0.5874 | 0.7905 | Pm | Monoclinic | 0.274 | 4.4516 |
S35 |
0.5922 | 0.5922 | 0.7867 | Amm2 | Orthorhombic | 0.275 | 4.4451 |
S41 |
0.5731 | 0.5721 | 0.8300 | Pm | Monoclinic | 0.272 | 4.2369 |
S42 |
0.5737 | 0.5737 | 0.8244 | Amm2 | Orthorhombic | 0.271 | 4.2491 |
S43 |
0.5746 | 0.5745 | 0.8220 | Amm2 | Orthorhombic | 0.271 | 4.2489 |
S44 |
0.5767 | 0.5689 | 0.8278 | Pm | Monoclinic | 0.272 | 4.2447 |
S45 |
0.5761 | 0.5761 | 0.8167 | Amm2 | Orthorhombic | 0.271 | 4.2530 |
式中, Et表示晶胞体系的总能量; ETi, EAl, ECe和EV分别为各元素在单质情况、 完全弛豫状态下的单原子能量; m, n, k和l分别代表在晶胞内各个元素的原子数; N代表晶胞体系的总原子数量。 经过计算后, Ti, Al, Ce和V的单原子能量分别为-1603.1183, -56.396, -1061.38和-1976.44 eV。 优化后纯γ-TiAl, Ce和V双掺杂γ-TiAl体系的总能量和平均形成能如表2所示。
计算结果显示, 各个双掺杂体系的Eb均为负值, 这表明它们具有较好的能量稳定性。 从表2可看出, 双掺杂体系的平均形成能较纯γ-TiAl体系有所提高, 因此体系结构稳定性也有不同程度的降低。对比Ce和V双掺杂体系的平均形成能,Ce和V同时替代Ti的体系比其他双掺杂体系平均形成能低。这说明双掺杂时,Ce和V原子更倾向于取代Ti原子,主要是因为Ce和V的共价半径与Ti的更接近。同时还可发现Ce替位Al后引起较大的晶格畸变,主要是因为Ce的共价半径(0.165 nm)和Al(0.118 nm)的相差较大。且在这些双掺杂体系中,S45体系的稳定性最好,说明掺杂时Ce和V原子极有可能分布在(001)面的Ti层上。
表2 Ce和V双掺杂γ-TiAl基合金体系的能量性质
Table 2 Energy properties of Ce and V co-doping γ-TiAl based alloy systems
Systems |
Energy properties |
|
Et/eV |
Eb/eV | |
S0 |
-13283.2833 | -0.4481 |
S1 |
-14658.8815 | -0.2784 |
S2 |
-14659.7057 | -0.3299 |
S31 |
-16205.1412 | -0.2495 |
S32 |
-16205.7835 | -0.2897 |
S33 |
-16205.5577 | -0.2756 |
S34 |
-16204.9527 | -0.2377 |
S35 |
-16204.7556 | -0.2254 |
S41 |
-13113.2454 | -0.3463 |
S42 |
-13113.2838 | -0.3487 |
S43 |
-13113.2626 | -0.3474 |
S44 |
-13113.2731 | -0.3481 |
S45 |
13113.5591 | -0.3659 |
在实际应用中, 材料必须具备力学稳定性, 即各个体系的弹性常数矩阵分量Cij (i,j=1, 2, 3, …, 6)需要满足Born-Huang判据
2.3 弹性模量和塑性
这些双掺杂体系共分为四方、 正交和单斜这3种晶系, 其弹性模量与弹性常数矩阵分量的关系也不尽相同
B=(2C11+2C12+4C13+C33)/9 (2)
G=(2C11-C12-2C13+C33+6C44+3C66)/15 (3)
而对正交晶系和单斜晶系, B和G与弹性常数的关系分别可表示为:
B=[C11+C22+C33+2(C12+C13+C23)]/9 (4)
G=[C11+C22+C33+3(C44+C55+C66)-C12-C13-C23)]/15 (5)
根据公式(2)~(5), 计算得到各个体系的B, G和B/G, 参见表3。
从表3中可以看出, 双掺杂体系的B变化在10%以内; 相对地G均明显减小, 表明其抗剪切应变能力明显减弱。 为便于分析, 绘制各体系弹性模量比B/G于图3中。
Pugh
表3 Ce和V双掺杂γ-TiAl基合金体系的弹性模量及其比值
Table 3 Elastic modulu and their ratios of Ce and V co-doping γ-TiAl based alloy systems
Systems |
B/GPa | G/GPa | B/G |
S0 |
110.3688 | 82.5593 | 1.3368 |
S1 |
101.5644 | 59.7496 | 1.6998 |
S2 |
101.7465 | 62.4579 | 1.6290 |
S31 |
115.8411 | 72.3268 | 1.8400 |
S32 |
100.9873 | 58.3259 | 1.6139 |
S33 |
117.0147 | 63.7160 | 1.6739 |
S34 |
119.1870 | 64.0991 | 1.6708 |
S35 |
100.8750 | 60.9155 | 1.7980 |
S41 |
102.6779 | 58.1125 | 1.6560 |
S42 |
103.7791 | 54.1176 | 1.7669 |
S43 |
103.3521 | 60.9291 | 1.9177 |
S44 |
100.8931 | 65.6001 | 1.6963 |
S45 |
102.3304 | 61.5907 | 1.6615 |
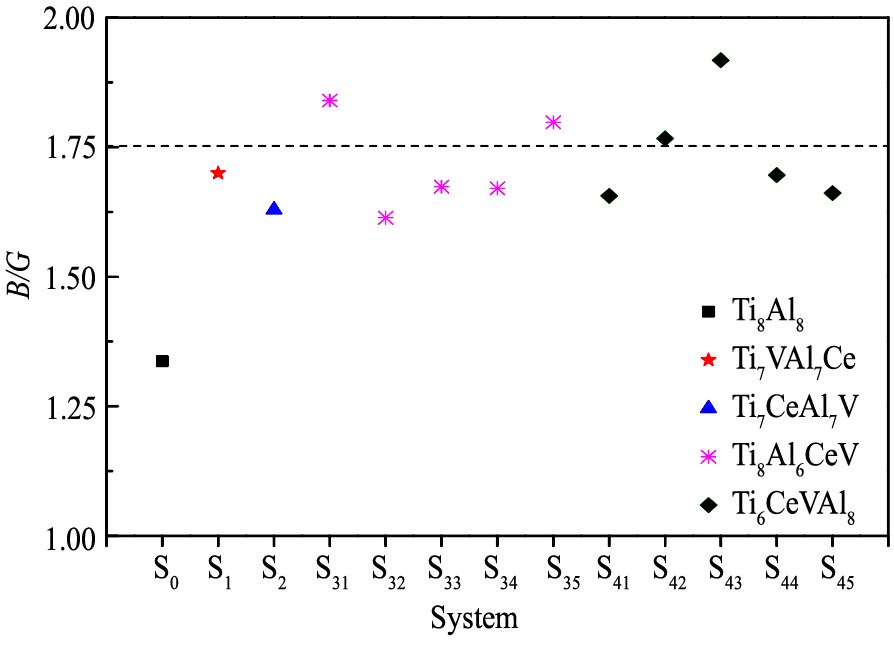
图3 Ce和V双掺杂γ-TiAl基合金体系的B/G
Fig.3 B/G of Ce and V co-doping γ-TiAl based alloy systems
2.4 电子性质和塑性
为了研究双掺杂改善γ-TiAl基合金塑性的微观机制, 绘出Ce和V双掺杂体系S32和S43, 以及纯γ-TiAl体系S0的总电子态密度DOS(density of states), 如图4所示。 由图4可知, 费米能级处的电子态密度均远大于零, 这表明掺杂前后体系均具有明显的电子导电性。 理论上, 体系的费米能级处的总DOS越小, 材料结构越稳定
计算三个体系的赝能隙列于表4。 研究表明, 赝能隙越宽, 原子结合的共价性越强
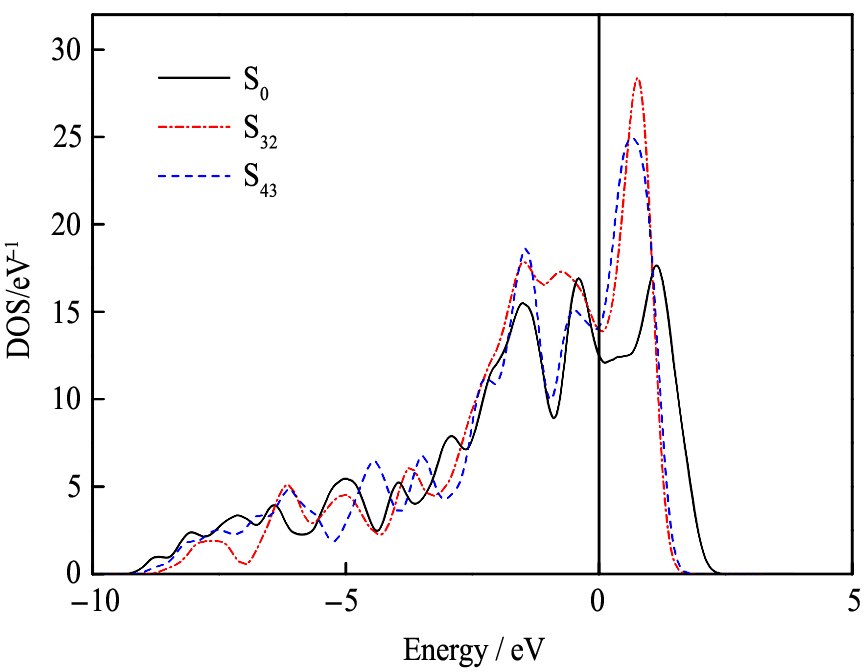
图4 Ce和V双掺杂及纯γ-TiAl体系的总态密度
Fig.4 DOS of Ce and V co-doping and pure γ-TiAl systems
表4 Ce和V双掺杂及纯γ-TiAl体系的赝能隙
Table 4 Pseudogaps of Ce and V co-doping and pure γ-TiAl systems
Systems |
Left peak/eV | Right peak/eV | Pseudogap/eV |
S0 |
-0.4117 | 1.1379 | 1.5496 |
S32 |
-0.7051 | 0.7619 | 1.4670 |
S43 |
-0.5040 | 0.6598 | 1.1638 |
为了进一步了解典型体系的电子性质、 电子分布与塑性的关联性, 本文计算了各体系的总态密度和分波态密度(PDOS, partial density of states)。 图5(a)~(c)分别表示体系S0, S32和S43在费米能级附近的各原子的态密度、 分波态密度及总态密度, 其中s, p, d, f分别代表电子轨道。
由图5(a)可知, 体系S0中, 在-8~2 eV的区域内Al-3p和Ti-3d电子呈现明显的交叠。 这种交叠表明: 纯γ-TiAl体系中Al-3p和Ti-3d电子之间存在较强的共价结合。 因共价键方向性较强, 在外加应力作用下材料易沿着原子结合能力弱的方向发生脆性断裂, 导致室温下纯γ-TiAl体系塑性较差
图5(b)和(c)显示, Ce和V替位掺杂后, 在费米能级附近Al-3p电子的态密度基本不变, 而Ti-3d电子的态密度明显降低, 并且Al-3p和Ti-3d电子交叠范围显著减少。 这说明Al-3p与Ti-3d电子杂化作用减弱, Ti和Al原子间的共价作用明显变弱、 金属键增强。
2.5 双掺杂体系中的化学键性质
材料的宏观性质取决于其微观结构。 这其中原子间的化学键性质起着关键作用。 原子间的重叠布居数可以较好地描述原子间化学键性质。 重叠布居数为正值标志为共价结合, 且量值越大共价键越强; 重叠布居数为负值标志原子间形成反键, 没有共价结合特征
本文以典型的Ce和V替位双掺杂体系S32和S0为代表, 计算了化学键的平均重叠布居数, 如表5所示。 从表5中可以看出, 对于体系S0, 其中Al-Al键共价性较强, 重叠布居数较大, 为0.72; Ti-Ti键的较弱, 重叠布居数为0.11; 而Al-Ti键更弱, 重叠布居数仅为0.07。 各个原子间结合均为共价键, 并且化学键具有显著的方向性。
对体系S32, Ce和V均替代Al原子, 掺杂的效果使其中Al-Al键的共价强度减弱, 重叠布居数减小为0.54; 相反, Ti-Ti键的共价强度显著升高, 重叠布居数达到0.43, Al-Ti键的共价强度也明显增大, 重叠布居数达到0.14。 综合而言, 化学键的各向异性程度明显降低。 而杂质原子Ce(或V)与其近邻原子间的化学键性质则变化多样。 Mulliken布居数计算结果表明, Ce原子和V原子失去部分电子, 而Al原子和Ti原子夺得部分电子。 Al-V键与Al-Al键的共价强度相当; Al-Ce键、 Ti-V键和Ti-Ce键则均成为反键(重叠布居数为负值), 不再具有共价键性质, 或者说这3个键已经变成非共价键。因此,杂质原子Ce和V已经向离子实转变,其附近区域已经具有金属性质。综合而言,这说明双替位掺杂后,原子间的共价键强度降低,金属键成分增加。
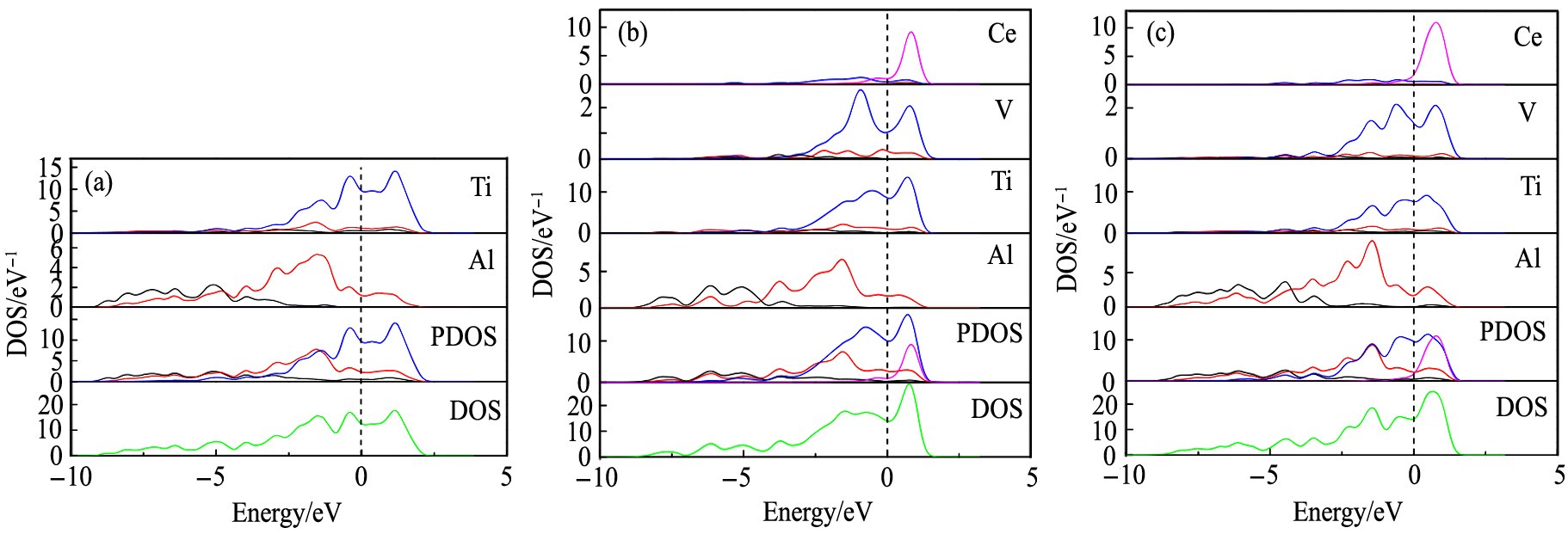
图5 各体系在费米能级附近的态密度
Fig.5 DOS near Fermi level of systems
(a)S0;(b)S32;(c)S43
表5 S0体系和S32体系中的平均重叠布居数 下载原图
Table 5 Average overlap populations of systems S0and S32
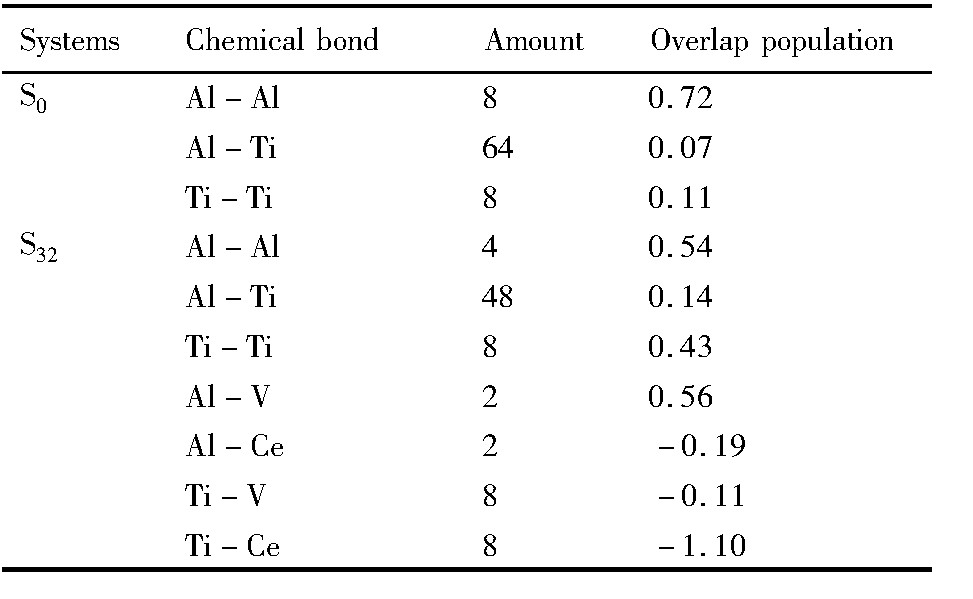
表5 S0体系和S32体系中的平均重叠布居数
图6和7给出了体系S0和S32中Ce原子和V原子所在(001)面和(100)面上的电荷密度。 从图6可以看出, 沿ab面(即(001)面)掺杂原子Ce和V与最近邻的Al原子之间的区域电荷密度降低,电荷分布方向性弱化。图7显示,Ce原子与同一ab平面内的Al原子间的电荷密度似乎有所加强,化学键性质不明显;较为明显的是,它与相邻ab面的Al原子间的电荷密度明显降低。对于Al-Al键,相邻ab面上的Al原子之间的电荷密度显著降低;同一ab面内的Al原子之间的电荷密度也明显降低。综合而言,Ce和V原子与其周围Al原子间的共价结合强度减弱,Al原子之间的共价结合强度也减弱。这一结果与重叠布居数是一致的。
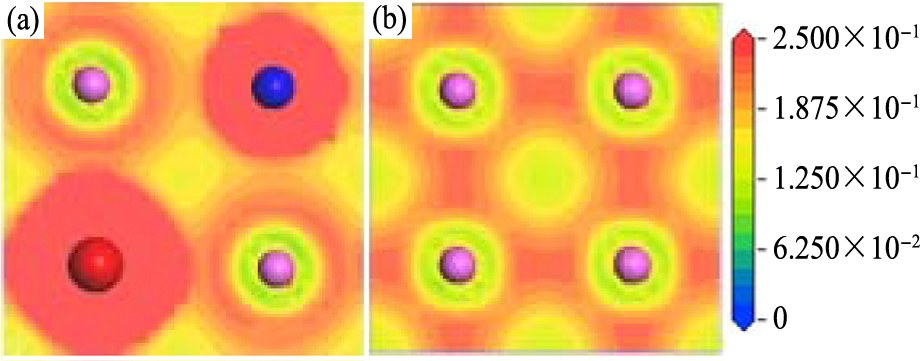
图6 体系S32和S0中(001)面的电荷密度
Fig.6 Charge densities of (001) plane in systems (a being in horizontal direction)
(a)S32;(b)S0
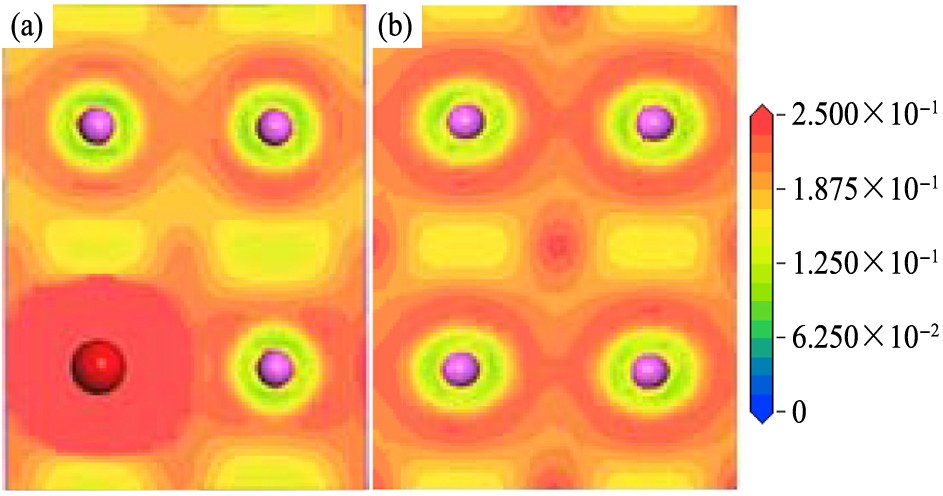
图7 体系S32和S0中(100)面的电荷密度
Fig.7 Charge densities of (100) plane of systems (b being in horizontal direction)
(a)S32;(b)S0
为了进一步研究Ce和V双替位掺杂体系中化学键的特性, 本文计算了体系S32和S0中(001)面上的差分电荷密度, 如图8所示。 由图可知, Ce和V替代Al双掺杂显著改变了Al原子周围电子排布形态, 电荷分布明显向各向同性转变。 这有利于材料塑性的改善。 Kenki等
Morrinaga等
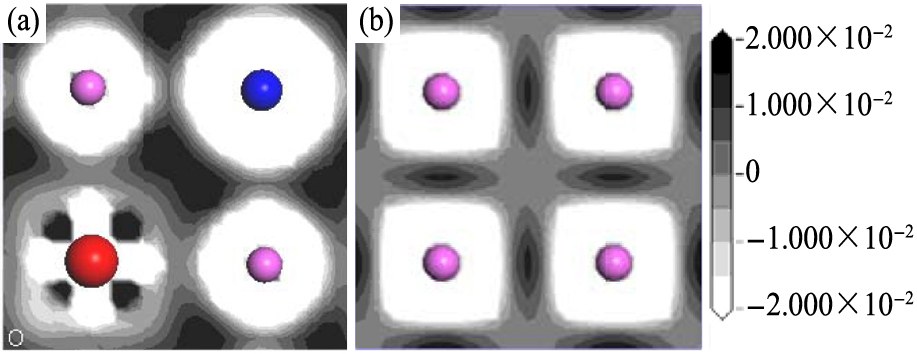
图8 S32和S0体系(001)面的差分电荷密度图
Fig.8 Charge density difference maps of (001) plane of system S32 and S0
(a)S32;(b)S0
3 结 论
通过以上对Ce和V原子双掺杂γ-TiAl基合金体系的能量、 塑性、 电子结构和化学键的计算与理论分析, 可以得出以下结论:
1. Ce和V元素替位掺杂γ-TiAl基合金后, 双掺杂体系的总能量和平均形成能均为负值, 说明双掺杂体系具有能量稳定性; 由各个体系满足Born-Huang判据, 表明各体系具有力学稳定性, 可以由实验制备并能稳定存在。 Ce和V原子同时替代Ti原子时体系的平均形成能较低, 说明Ce和V原子更倾向于替代Ti原子。
2. 通过比较掺杂前后体系的弹性模量比B/G发现, 体系S32, S35, S42和S43等的塑性均有明显的改善趋势。 与此相应, 电子态密度显示, Ce和V原子双掺杂后, Ti-3d轨道电子的态密度主峰强度变弱, Al-3p与Ti-3d电子杂化作用减弱, 同时双掺杂体系的赝能隙减小。
3. 对体系S32, 化学键重叠布居数和电荷密度显示, Ce和V元素替位Al, 其塑性改善的内在机制是: 化学键Al-p-Ti-d的共价结合强度显著弱化; Ti-d-Ti-d键显著增强; Al-Al键明显弱化。
参考文献

