文章编号: 1004-0609(2005)07-1069-06
Al-Li合金时效初期的价键分析
高英俊1, 2, 黄创高1, 莫其逢1, 蓝志强1, 刘 慧1, 韦银燕1
(1. 广西大学 物理科学与工程技术学院, 南宁 530004; 2. 中国科学院 国际材料物理中心, 沈阳 110016)
摘要: 运用固体经验电子理论(EET), 对Al-Li合金时效初期的若干偏聚晶胞的价电子结构进行了计算。 计算结果表明: 不包含空位的偏聚晶胞的键络最强键为Al―Al键, 其中Al原子的共价半径较Li原子的共价半径要大; 而含空位的偏聚晶胞的最强键为Al―Li键, Al原子的共价半径要比Li原子的共价半径要小; 在空位存在的情况下, 由于Al原子与Li原子的电负性相差明显, 促使Al和Li原子结合, 倾向形成Al-Li短程序结构偏聚区, 这种含空位的短程序结构很可能就是δ′(Al3Li)亚稳相的前兆结构和生长胚胎; 由于Al-Li-空位有序偏聚晶胞的Al―Li键络比基体键络要强许多, 因此, 淬火过程中合金生成的Al-Li-空位偏聚晶胞对合金过饱和固溶体起主要强化作用; 后续析出的δ′(Al3Li)亚稳相键络各项异性显著, 键络强度明显提高; 由于Al3Li与基体共格, 其大量均匀弥散析出起到提升基体整体键络强度, 同样对合金产生强化作用。
关键词: Al-Li合金; Al3Li; 空位; 价电子结构; 力学性能 中图分类号: TG111.1
文献标识码: A
Calculation on valence electronic structures of Al-Li alloy under earlier aging condition
GAO Ying-jun1, 2, HUANG Chuang-gao1, MO Qi-feng1, LAN Zhi-qiang1, LIU Hui1, WEI Yin-yan1
(1. School of Physics Science and Technology, Guangxi University, Nanning 530004, China;
2. Center of International Materials and Physics, Chinese Academy of Sciences, Shenyang 110016, China)
Abstract: The valence electron structures of the segregated cell of Al-Li alloy in earlier aging condition were calculated according to the empirical electron theory (EET) in solid. The results show: the strongest bond is the Al-Al bond in the segregated cell without containing vacancy, where the Al atomic covalence radius is greater than that of Li atom in the cell; while the strongest bond is the Al-Li bond in the segregated cell containing vacancy, and the Al atomic covalence radius in the cell is less than that of Li atom. Since the difference of electronagativity between the Al and Li atoms is obvious, it is inclined to formed the Al-Li segregated cell of short range order structure in the condition of vacancy present. The short range order structure containing vacancy is probably the embryo or precursor structure of the metastable phase δ′(Al3Li). Because the strongest covalent bond in the Al-Li-vacancy segregated cell in alloy formed in quenching is the main strength reason for supersaturated solid solution of alloy. The bond net of succeeding precipitation of δ′(Al3Li) has the picture of anisotropic Al-Al bonding and the bond intensity enhanced. Since the δ′(Al3Li) is coherence with matrix, the bond net strength is enhanced by the precipitation of δ′(Al3Li) and so strengthen the alloy.
Key words: Al-Li alloy; Al3Li; vacancy; covalence bond; mechanical properties
Al-Li合金具有低密度, 高比强度和比模量, 良好的耐蚀性和优异的低温特性等特点[1-3], 已成为航空航天的重要结构材料。 对于经固溶淬火时效的Al-Li合金, 通常认为沉淀惯序是先析出与基体共格的δ′(Al3Li)亚稳相, 随着时效时间的延长, 最后析出稳定的δ(AlLi)平衡相。 但最近几年, 实验研究[4-7]发现Li含量大于5.5%(摩尔分数)的Al-Li合金固溶淬火, 得到的过饱和固溶体, 在早期时效阶段析出亚稳δ′(Al3Li)相之前, 在极短时间内合金经过协同有序化和Spinodal分解等相变过程[8, 9], 首先形成类似G.P区或溶质原子短程有序的前兆溶质原子偏聚结构区, 然后逐渐演化形成有序的δ′(Al3Li)亚稳相。 一些文献[10-14]还报道了应用电阻测量方法和低场Hall效应方法测量淬火态合金的溶质原子和空位对合金的电阻和Hall系数的影响, 揭示了溶质原子与空位对时效中δ′相形成的作用。 这些前兆原子偏聚结构的演化是受过剩淬火空位的控制[6,15], 空位的状态对这些原子偏聚区结构的形成生长产生重要的作用。 实验研究[2]还表明, Al-Li合金所具有的高弹性模量是与Li原子及其周围原子成键的价电子结构有着紧密的联系, 这也进一步说明了时效过程中由于Al和Li原子的电负性差而生成的化合物微观相结构, 改变了合金内部的原子成键状态, 从而引起合金性能发生改变。 现在, 人们更注重从价电子结构层次揭示合金具有的优良宏观性能的内在原因。
近些年国内外都有学者从理论和实验上对Al-Li合金的价电子结构进行了研究[16, 17]。 尽管运用第一原理方法对Al-Li合金的价电子结构计算, 取得了有意义的结果, 但计算过程较为复杂。 基于价键理论[18]和能带理论建立的固体经验电子理论(EET)[19], 则提供了一个处理复杂体系价电子结构的简捷计算方法――键距差 (BLD)法, 成功地用于合金的原子偏聚与合金相变研究[20], 使得研究合金的宏观性能可以追溯到合金原子的价电子结构层次, 为合金改性设计提供了深层次的理论指导[20, 21]。 本文作者运用EET理论, 计算Al-Li固溶淬火态合金时效初期合金内部原子的价电子成键状态, 从价电子结构层次分析Al-Li合金时效对性能的影响。
1 晶胞模型
对于Al-Li合金相图富Al端的α相经高温固溶冰水淬火后, 将形成过饱和的Li溶质固溶体。 与此同时, 由于Li原子与空位有强的结合作用, 在固溶体中将会形成大量的过饱和空位缺陷。 在Al-Li合金中Li含量不高(4%〈x〈12%)的情况下, 考虑到Al与Li原子之间具有大的化合价之差而引起的化学亲合作用, 同时Li原子对空位有较强的俘获能力[11], 因此, 在Al-Li合金固溶处理过程中将会出现局部微观不均匀性。 对Al-Li合金固溶体的微观不均匀性可利用“混合偏聚晶胞”模型[20]近似描述。 这种模型认为, 在合金固溶状态, Al-Li固溶体可看成由几类偏聚晶胞混合堆砌构成: 1) 纯Al晶胞(FCC晶胞); 2) 含Li的Al晶胞(FCC晶胞), 即Li原子替代了纯Al晶胞的部分面心Al原子; 3) 考虑到合金固溶处理过程中, 就有大量的非平衡空位生成, 而Li原子与空位又有很强的结合能力[11], 这些过剩空位被Li原子俘获, 易形成Li原子-空位对, 所以Al-Li固溶体中还应该包含有Li原子与空位相互作用所形成的Al-Li-空位的偏聚晶胞。 这几种偏聚晶胞经合金的固溶淬火, 都可被淬火态合金保存下来, 这些微结构将对合金的性能产生重要的影响。
为研究和计算方便, 并能与文献[2, 6]中的实验结果进行比较, 这里以Al-8%Li(摩尔分数)合金作为研究对象。 对于这种Li浓度的合金, 可以粗略的估算出含Li的Al-Li偏聚晶胞相隔的平均距离大约为2~3个原子间距。 在熔点温度(875K)附近的Al合金固溶体, 文献[22]给出其包含的空位浓度可达10-3。 此时, 可以粗略的估计出Al-Li固溶体中空位的平均间距在10个原子的距离左右。 因此, 可以近似认为Al-Li-空位晶胞之间的平均距离为6~7个原子间距。 因此可见, 对所研究的Al-8%Li合金而言, 含空位的Al-Li-空位的偏聚晶胞的数量要比不含空位的Al-Li的偏聚晶胞数量相对要少。 按照文献[6]指出的Al-Li淬火合金是协同有序化的, 因此, 这里仅考虑具有对称性晶胞情况是合理的。 对于不含空位的Al-Li晶胞, 为研究方便起见只考虑三种不同成分类型的对称性晶胞, 例如含Li分别为6.25%, 12.5%, 25%(摩尔分数)的Al-Li晶胞。 这3种含不同Li成分的晶胞分别用Al-6.25%Li、 Al-12.5%Li、 Al-25%Li表示, 如图1(c), 图2(b), 图1(b)所示。 考虑到Li原子与空位有较强的结合能, 易形成Li原子-空位对, 故在含空位的Al-Li-空位复合偏聚晶胞中, Li原子和空位应尽量占据面心的位置, 以便使Li原子与空位位置尽量地靠近。 选取这几种对称晶胞结构模型作为研究对象, 主要是依据文献[14]给出的Al-Li合金高分辨电镜照片。 该照片显示出合金淬火态的前兆结构具有短程有序结构近似为L12型结构, 但热稳定性不如Al3Li晶体结构好, 并且这些前兆结构与基体的边界[8, 9]较模糊。 这些模糊的边界可能是由于存在空位团、 位错、 位错环等缺陷结构。 这里考虑的Al-Li-空位晶胞具有面心立方结构如图2(a)所示。
按照文献[22]指出的铝晶体点阵常数随Li的增加而下降, 这说明了含Li的偏聚晶胞的晶格常数小于不含Li的纯Al晶胞的晶格常数。 实验测得的点阵常数为这两种晶胞尺寸的计权平均值。 已知纯Al的晶格常数[22]为a0 =0.40496nm, 由上述实验规律可以推出含8%Li的Al-Li合金的点阵常数为a=0.4046nm, 即Al-Li固溶体中, 当摩尔分数为8%时, 平均晶格常数为a[TX-]= 0.4046nm, 则容易计算出含Li的Al-Li偏聚晶胞的晶格常数为a1=0.40434nm。 为计算方便起见, Al-Li-空位的偏聚晶胞的晶格常数近似地取Al-Li偏聚晶胞的晶格常数代替, 而将空位对晶格常数的影响通过Al和Li原子的杂阶变化来反映。 在固溶体淬火时效过程中, 考虑到还会析出与基体共格的亚稳δ′(Al3Li)相, 该析出相具有L12型晶体结构, 其晶格常数[22]为a=0.4010nm。 δ′(Al3Li)相的晶体结构如图2(c)所示。
2 计算方法与结果
固体中原子的价电子结构在这里是指该固体中原子所处的状态以及原子形成共价键的键络分布。 按照EET[19]理论, 原子的共价电子是分布在连接最近邻、 次近邻, 以及s近邻原子的键上。 各键上共价电子对数(即键级nα)由下列原子键距公式表示:
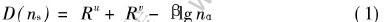
式中 R为原子单键半径; 参数β的数值按文献[19]中的式(3)~(14)确定。 晶胞内的共价电子数可以写为

式中 k1、 k2分别为晶胞中u、 v原子的个数; nuc、 nvc分别为u、 v原子的共价电子数; Iαs为nα键级的等同键数, 各等同键数的选取可依照文献[19]给出的方法来确定。 由于各晶胞的结构已确定, 实验晶[CM(22]格常数在文献[22]中已给出, 因此, 运用键距差方法[19]建立最强键nA方程, 并参见文献[20, 23-26]的求解步骤, 联立(1)、 (2)等方程组, 逐个计算各晶胞中原子成键的价电子结构, 并利用BLD判据确定原子的杂阶状态。 计算得到的各晶胞的共价键结果列于表1~6。 表中的σ和K分别表示原子的杂阶状态和空位。

图1 不同Li含量的Al-Li晶胞
Fig.1 Al-Li cell with different Li contents

图2 Al-Li-空位晶胞面心立方结构
Fig.2 FCC structures of Al-Li-vacancy cells
表1 纯Al晶胞的共价键强度
Table 1 Covalent bonds of pure Al cell

表2 Al-25%Li晶胞的共价键强度
Table 2 Covalent bonds of Al-25%Li cell
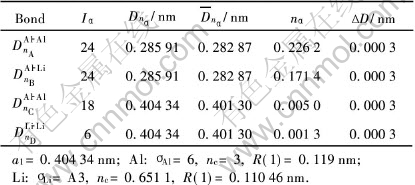
表3 Al-6.25%Li晶胞的共价键强度
Table 3 Covalent bonds of Al-6.25%Li cell

表4 Al-Li-□晶胞的共价键强度
Table 4 Covalent bonds of Al-Li-□ cell

表5 Al-12.5%Li晶胞的共价键强度
Table 5 Covalent bonds of Al-12.5%Li cell

表6 Al3Li相单位晶胞的共价键强度
Table 6 Covalent bonds of Al3Li cell
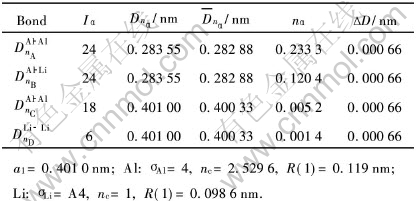
3 分析与讨论
由表2、 3、 5可见, 计算的3种不含空位的Al-Li偏聚晶胞中的最强共价键都为Al―Al键, 但Li含量为12.5%和25%的晶胞键络骨架最强键(nA=0.2250~0.2260)要比纯Al晶胞的键络骨架最强键[23](nA=0.2086)要强。 而含6.25%Li的晶胞键络骨架则与纯Al晶胞键络骨架相当, 两者的最强Al―Al键nA几乎相等, 约为0.2075~0.2085。 这表明Li含量较低时, Al-Li偏聚晶胞对合金键络增强不明显。 由表4可见, 对于含空位的Al-Li晶胞, 最强共价键已不再是Al―Al键, 而是转变成Al―Li键, 这表明淬火过程中由于空位进入晶胞中, 使得Al与Li的结合倾向增强。 实验[11, 12] 观测到在Al-Li合金淬火态就已存在Al-Li短程有序结构, 这也说明由于Li原子与空位有较强的结合倾向, 过剩空位的存在有利于形成Al-Li短程有序结构。 文献[2]指出Al-Li合金淬火态的弹性模量随Li含量增加而增加, 利用这种Al-Li-空位偏聚晶胞的存在, 就很容易对这一实验结果进行合理的解释。 由于Li含量的增加, Li原子俘获的空位数也增加, 因此, 淬火过程形成的Al-Li-空位的偏聚晶胞的数量也增加, 这些Al―Li键络骨架较强的偏聚晶胞, 就起到提升淬火态合金的弹性模量。
随着时效温度升高和时效时间延长, 合金中空位被激活, 引起Al-Li-空位偏聚晶胞中空位附近的键络骨架折断解体, 附近的溶质原子重组。 这一过程对应于文献[6]给出的DSC分析曲线中, 处于形成结构的吸热峰前的另一个明显的吸热峰, 但这个峰较宽。 这里的Al-Li-空位偏聚晶胞很可能就是文献[5, 6]指出的在合金淬火时发生的协同有序和Spinodal分解的产物, 这些有序结构区域很可能将成为文献[6]中指出的淬火时效初期形成δ′(Al3Li)相的前兆结构或胚胎结构。 文献[20]还报道了这些偏聚前兆结构周围存在较多的空位团, 位错环等缺陷。 这些都表明前兆偏聚结构通常是伴随有空位缺陷的结构。 由于空位的存在, 使得合金溶质原子经过扩散更容易形成短程有序化结构。
随着时效时间进一步延长, 亚稳的相逐渐析出。 由表6可见, δ′的最强共价键为Al―Al键, 强度达到nA=0.2333, 比Al-Li-空位偏聚晶胞的最强键络(nA=0.22484)有所提高, 但次强键较弱, 仅为nA=0.1244。 这也表明δ′(Al3Li)的键络结构各向异性明显, 这与文献[17]给出的计算结果是相一致的。 由于δ′的键络骨架比合金基体的键络骨架更坚实, 并且与基体界面有较好的共格关系。 因此, 大量的δ′析出能够在固溶强化的基础上明显增强基体合金的键络结构, 提高合金的弹性模量, 同时也使合金的强度有较大的提高。
由表2的结果可见, 对于含Li为25%的晶胞, Al原子的状态处于最高的第6杂阶, 最外层3个价电子都转化为共价电子, 无导电的自由电子, 而实验给出的导电率并没有明显下降, 因此这种原子状态的晶胞实际出现的可能性很小。 这种情形如果出现, 说明淬火过程可能引起Al原子状态发生很大的变化, 其原因可能是合金内部的原子相互作用很强, 内应力很大的缘故。 通常情况下, Al原子处于这种完全共价电子状态是不易出现的。
由表3和表5可见, 对于不含空位的Al-Li晶胞中, Al原子的杂阶处于第4和5杂阶, Li原子处于第3杂阶。 含Li为6.25%的晶胞键强相对较弱, 不能对基体起到强化作用。 而Li含量较高的如12.5%的晶胞, 键络较强, 在淬火态合金中能起到强化作用。
由表4的结果还可以看到, Al-Li-空位晶胞中Li原子处于第2杂阶, 其共价半径为0.1240nm, 比Al原子的共价半径0.119nm要大, 最强键为Al―Li键, 这表明由于Al和Li原子的电负性相差较大, 空位的存在, 使得Li原子的体积扩展, 有向Al原子转移电荷的倾向, 容易形成Al-Li原子偏聚状态, 即固溶淬火过程中存在向Al-Li有序化方向发展趋势。 由于空位作用引起的Al―Li键比Al―Al键要强, 使得带空位的Al-Li偏聚晶胞在淬火过程中极容易生成, 从而对淬火态合金有一定的强化作用。 而不含空位的Al-Li偏聚晶胞中的Li原子则处于第3杂阶, 其共价半径为0.1144nm, 都比Al原子的半径0.119nm要小。 这种不含空位的晶胞的Li原子由于半径较小, 时效过程中容易通过空位扩散移动。 而Al-Li-空位晶胞中, Li原子半径较大, 与Al原子的共价结合较强, 因而难以激活成为自由原子而扩散运动, 而需要较高的温度才能破坏Al―Li键络。
以上计算的几种可能的晶胞结构中, 固溶淬火中最可能形成的偏聚晶胞为Al-12.5%Li和Al-Li-空位偏聚晶胞, 它们都可能对固溶淬火合金起强化作用。
4 结论
1) 含空位的Al-Li晶胞键络最强, 最强共价键为Al―Li键。 晶胞中的Li原子半径比Al原子半径大是由于空位存在, 使得Li原子外层电子运动范围有所扩展, 原子成键区域扩大引起的。 Li原子半径较大使得Li原子扩散迁移较难。 Al-Li-空位偏聚晶胞很可能就是淬火时效初期形成δ′(Al3Li)相的前兆结构或胚胎结构。
2) 不含空位的晶胞, 最强键为Al―Al键, Li原子半径较小, 易于沿过剩空位运动。 Li含量低的晶胞键络强度较弱, 对合金基体强化作用不明显。 含12.5%Li的Al-Li晶胞, 键络较强, 在淬火态合金中能起到强化作用。 Li含量为25%的晶胞, 由于要求Al原子外层3个价电子都为共价电子, 这种情况一般情况下难以实现, 故这种偏聚晶胞出现的可能性很小。
3) δ′(Al3Li)相键络各项异性显著, 其最强共价键比Al-Li偏聚晶胞的最强共价键和基体的共价键络要强, 这是其对合金基体其强化作用的内在原因。
REFERENCES
[1]Lavernia E J, Srivatsan T S. Review of strength and fracture behavior and ductility of Al-Li alloys[J]. J Mater Sci, 1990, 25: 1137-1158.
[2]Noble B, Harris S J, Dinsdale K. The elastic modulus of Al-Li alloys[J]. J Mater Sci, 1982, 17: 461-468.
[3]Enrique J, Nicholas J. Review aluminum-lithium alloys[J]. J Mater Sci, 1987, 22: 1521-1529.
[4]Kassab T, Menand A, Chambreland S. The early stage of decomposition of Al-Li alloys[J]. Surface Science, 1992, 266: 333-336.
[5]Wei Y H, Wang S T. Experiment evidence for spinodal decomposition in Al-12.7%Li alloys[J]. Material Letter, 1996, 28: 123-127.
[6]Noble B. Evidence for pre β′ precipitation events in an Al-Li alloys[J]. Scripta Metall, 1995, 33(1): 33-37.
[7]Meng F L, Chai Z G, Li J X. Small-angle X-ray scattering study of the β′ phase in Al-Li alloys[J]. Mater Characterization, 2001, 47: 43-46.
[8]Noble B, Bray S E. The interfacial energy of β′ precipitation in Al-Li alloy[J]. Mater Sci Eng A, 1999, A266: 80-85.
[9]Perez J I, Madariaga G. Quantitative analysis of β′ precipitation kinetics in Al-Li alloys[J]. Acta Mater, 2000, 48(4): 1283-1296.
[10]Boukos N, Papastaikoudis C. The influence of β′ precipitates on the electrical resistivity and low-field Hall coefficient of Al-Li alloys[J]. Phil Mag B, 1994, 70(1): 67-75.
[11]Ceresara S, Giarda A, Sanchez A. Annealing of vacancies and ageing in Al-Li alloys[J]. Phil Mag, 1977, 35(1): 97-110.
[12]Gregson P J, Flower H M. Role of vacancies in coprecipitation of δ′- and S-phase in Al-Li-Cu-Mg alloys[J]. Mater Sci Tech, 1986, 2: 349-353.
[13]Gaber A, Afify N. Characterization of the precipitates in Al-Li alloy using thermal measurements and TEM[J]. Physica B, 2002, 315(1): 1-6.
[14]Sato T, Tanhka N, Takahashi T. High resolution lattice images of ordered structure in Al-Li alloy[J]. Transactions of the JIM, 1988, 29(1): 17-25.
[15]Noble B, Bray S E. On the Al3Li metastable solvus in Al-Li alloy[J]. Acta Mater, 1998, 46(17): 6163-6171.
[16]Poduri R. Computer simulation of morphological evolution and coarsening kinetics of δ′(Al3Li) precipi-tation in Al-Li alloys[J]. Acta Mater, 1998, 46(11): 3915-3920.
[17]Guo X Q, Podloucky R, Xu J H. Cohesive and structural properties of Al3Li[J]. Phys Rev B, 1990, 46(18): 12432-12440.
[18]Pauling L. The Nature of the Chemical Bond[M]. San Simeon: Corrnell University Press, 1960. 300-400.
[19]张瑞林. 固体与分子经验电子理论[M]. 长春: 吉林科学技术出版社, 1993. 1-90.
ZHANG Rui-lin. The Empirical Electron Theory of Solids and Molecules[M]. Changchun: Jilin Science and Technology Press, 1993. 1-90.
[20]刘志林. 合金价电子结构与成分分析[M]. 长春: 吉林科学技术出版社, 1990. 1-300.
LIU Zhi-lin. The Valence Electron Structure and Composition Design of Alloys[M]. Changchun: Jilin Science and Technology Press, 1990. 1-300.
[21]Li Z L, Ma C X, Liu Z L. Valence electron structure of high property steel and its composition design[J]. Acta Metallurgica Sinica, 1999, 12(4): 408-416.
[22]Mondolfo L F. Structure and Propertys of Aluminum Alloys[M]. London: Butterworths Press, 1976. 200-400.
[23]高英俊, 韩永剑, 赵妙. Al-Zn固溶体价电子结构与Spinodal 分解现象[J]. 中国有色金属学报, 2004, 14(5): 730-735.
GAO Ying-jun, HAN Yong-jian, ZHAO Miao. Electron structure of Al-Zn solid solutions and spinodal decomposition[J]. The Chinese Journal of Nonferrous Metals, 2004, 14(5): 730-734.
[24]GAO Ying-jun, BAN Dong-mei, HAN Yong-jian. Atomic bonding and mechanical properties of Al-Mg-Zr-Sc alloy[J]. Trans Nonferrous Met Soc China, 2004, 14(5): 922-927.
[25]GAO Ying-jun, HAN Yong-jian. Electron structure and interface energy of GP zone in Al-Zn alloy[J]. Mater Sci Forum, 2005, 475-479: 3131-3135.
[26]高英俊, 钟夏平, 刘慧, 等. Al-Cu合金亚稳相的价电子结构分析[J]. 稀有金属, 2003, 27(6): 845-849.
GAO Ying-jun, ZHONG Xia-ping, LIU Hui. Calculation on valence electron structures of metastable phase in Al-Cu alloy[J]. The Chinese Journal of Rare Metals, 2003, 27(6): 845-849.
基金项目: 国家自然科学基金资助项目(50061001); 广西科学基金资助项目(桂科基0342004-1); 广西“十百千人才工程”资助项目(2001207)
收稿日期: 2004-11-29; 修订日期: 2005-04-06
作者简介: 高英俊(1962-), 男, 教授, 博士.
通讯作者: 高英俊, 教授; 电话: 0771-3236667; E-mail: Gaoyj@gxu.edu.cn
(编辑 陈爱华)

