���±��: 1004-0609(2006)07-1207-07
����Ԫ��Cu������ѧ����
�ջԽ�, л����, ���콨, ���, �����, ������, ��ҫׯ
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ��ɳ 410083)
ժ Ҫ: ����SGTE���������ݿ���Gibbs�ܵı���ʽ, ���JANAF����ѧʵ������, ����С���˷��Խ���Ԫ��Cu��Gibbs�ܱ���ʽ�������������� �����������ת�����ݡ� ת���ʺ�ת���طֱ�Ϊ-0.187J/(mol��K)�� 13.138kJ/mol��9.675J/(mol��K), ��ʵ�����ݷ��Ϻܺá� ����Debye-Gr��neisenģ���о�Cu�� 0K���۵������ѧ����, �������ģ�ͼ���ĵ�������ֵƫ��, �Ӷ�����ģ�ͼ����ת���غ�Gibbs��ƫ�� ����Debye-Gr��neisenģ���о�����Һ�������ѧ����, ��ͨ�������۵����ݡ� �ʺ���ֵ��������, �õ�Һ��Cu�����ݺ�Gibbs�ܵ�����ѧ����, �����ʵ��ֵ���ݷ��ϽϺá�
�ؼ���: Cu; Gibbs��; ����; ����ѧ����; Debye-Gr��neisenģ��; ����Һ��
��ͼ�����: TG111 ���ױ�ʶ��: A
Thermodynamic properties of pure elemental Cu
TAO Hui-jin, XIE You-qing, PENG Hong-jian, YU Fang-xin,
LIU Rui-feng, LI Xiao-bo, NIE Yao-zhuang
(School of Materials Science and Engineering, Central South University,
Changsha 410083, China)
Abstract: The transition data and Gibbs energy functions of pure elemental Cu in SGTE database were reassessed using the least-square method and adopting the newly available thermochemical reference JANAF data in the fourth edition. The results of transitional heat capacity, enthalpy and entropy are -0.187J/(mol��K), 13.138kJ/mol and 9.675J/(mol��K), respectively, and these data agree well with JANAF data. Debye-Gr��neisen model was applied to study the thermodynamic properties of fcc and supercooled liquid phase from 0K to 1357.77K. It is found that the heat capacity of Debye-Gr��neisen model at low temperature is lower than experimental data and leads to the higher transitional entropy and Gibbs energy. Keeping the continuum of cp, H and S between the supercooled and real liquid phase of Cu at melting point, the thermodynamic properties of liquid Cu can be obtained and they agree well with JANAF data as well.
Key words: Cu; Gibbs energy; assessment; thermodynamic properties; Debye-Gr��neisen model; supercooled liquid
����������ѧ���ݶ��ڶ���Ԫ����ѧ��ϵ��ģ�������������Ҫ, ����һ�����ݵ������кܶ��ַ���[1-4]�� SGTE(Scientific Group Thermodata Europe)���������ݿ�[5]�Ѿ�������298.15K����78��Ԫ�������������ѧ���ݡ� ����, �����ݿ��н���Ԫ��Cu�����������ʵ��ֵ���ϴ�, ��������������JANAF�����Ѿ��õ��˸���[6]�� ���, �б�Ҫ����������һ���ݿ���298.15K���ϵ�����ѧ������ ͬʱ, ��������ģ���о�����������Ƹ����ݿ�0~298.15K���¶ε�����ѧ���ݺ�������[7]��
�������߲���SGTE���������ݿ���Gibbs�ܵı�����ʽ, ���JANAF����ѧʵ������, ������С���˷�����������Gibbs�ܱ���ʽ�еIJ����� ����Debye-Gr��neisenģ��[8-10]�о��˹������Һ�������ѧ����, ͨ�������۵����ݡ� �ʺ���ֵ��������, ���SGTE�ķ���������Һ������ݺ���Ӧ����ѧ���ʡ� ͨ����SGTE���������ݿ����뱾�����������������������ģ�ͽ�����жԱ�, ���ֱ��������������Ľ����ʵ�������ܺܺ÷���, �ر��������ת�����ݡ� ת���ʺ�ת������ʵ��������ȫһ��, ֤���˱�����������������ĺ�����ɿ���, �Ӷ�Ϊ�Ͻ�����ѧƽ������ṩ���µĻ������ݡ�
1 ԭ���뷽��
1.1 Gibbs�ܵ�����ԭ���ͷ���
��ȷ������ѧ��������ʽ֮ǰ, ��Cu��ת�����ݽ��������������� ת����¶���Ȼ����ΪSGTE���ݿ��1357.77K, �������ݶ���Դ������[6], ������1���С�
��1 ��Ԫ��Cu��ת������
Table 1 Transition data of pure Cu

1.1.1 ��ѹ����
��SGTE���������ݿ���, ��ѹ����Ϊ�¶�T��-2�Ρ� 0�Ρ� 1�κ�2�η����������, ʵ���Ϻ�ѹ���ݱ���ʽҲ�����ؿ�Ϊ�¶�T��-2�� -1�� 0�� ���� 4�η���7�����[11]�� ����, SGTE���������ݿ����ȫ����о��������������ѧ��������, �����ڶ���ƽ������ѧ�ļ������������, ���Ա��ĵ�Gibbs��������Ȼ����SGTE���ݿ�ĺ�����ʽ��
1.1.2 ��
��SGTE���ݿ���ͬ, ��������ʽ��298.15K��105Pa�������ȶ�״̬Ԫ�ص���ֵHSERΪ�ο�̬�� ���ڽ���Cu, HSER=Hfcc298.15K, ���,
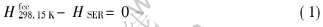
������һ��������ȷ��fcc�������ֵ����ʽ, ��ϱ�1��ת���ʦ�Htrans, ���Խ�һ���õ��۵�ʱҺ�����ֵ, ������һ��ֵ����ȷ��Һ����ֵ����ʽ�еIJ�����
1.1.3 ��
���ݱ�1��298.15K����S298.15, ���Եõ�

�ɴ˿���ȷ��fcc����ı���ʽ, ���ݱ�1���۵��¶�ʱ��ת����ֵ��Strans, ���Լ����۵�ʱҺ�����ֵ, ������һ��ֵ����ȷ��Һ����ֵ����ʽ�еIJ�����
1.1.4 ����Ȼ̬������ȷ��
��Ȼ̬��Һ��ı���ʽȷ����, ���öԹ��ȹ�������T-9��ͶԹ���Һ������T7��Ĵ�������, �����ֶַκ������۵���ֵ����ֵ��������, ����ȷ��SGTE���ݿ���298.15K���е�2843.261K���ȹ������Һ��ı���ʽ; ͨ������Saunders[2]�ľ����ȶ���������ȷ��������ṹ��Gibbs�ܱ���ʽ��
1.2 Debye-Gr��neisenģ���о�
����������Ԫ��Cu��Gibbs�ܱ���ʽ������������������, ����, ����0~298.15K������ѧ��Ϣ��Ȼ��δ֪��, �����б�Ҫ��SGTE���������ݿ�����ѧ��Ϣ���в��䡣 ���Ĺ���������ʵ��������ϵķ���, ����ֱ����Debye-Gr��neisenģ�ͽ��м��㡣 ��298.15K���۵���¶�����, ͬ��������ģ�ͼ���������Һ�������ѧ����, ���������ʵ�����ݽ��жԱȡ�
1.2.1 ����
��0K��̬����ԭ����Ϊ�ο�״̬, ��fcc�����0K�����Ec[12]����ȷ��0K����ֵ:

1.2.2 ����Һ��
���ǵ�Һ��Ķ̳�����, ����Һ���ڼ����е���fcc�ṹ������[15, 16], ���ٶ��°��¶���0K����ܵ�ƽ����������[17], ����ʽ�е�k��Q0��fcc������ͬ, ����Debye-Gr��neisenģ�ͽ�������ѧ�����о�, �����㷨���¡�
����298.15K SGTE���������ݿ��еľ����ȶ�������Gliquid-fccȷ������Һ���Gibbs��GliquidΪ

�����ļ��㷽������, ����Debye-Gr��neisenģ��, �����������cv����������ϵ���¡�, ����Һ�����Cu�۵�ʱ��ʵ���ܶ�ֵ[18], ����Ħ�����V���嵯��ģ��B, ������õ�����Һ��ĺ�������cp, �Ӷ����Խ���Gibbs�ܵ���������ѧ���ʵļ��㡣
1.2.3 Һ��
����Һ������ݵ�����ѧ����ȷ��֮��, ��SGTE�Ĵ���������ͬ, ���۵�ʱ������Ȼ̬Һ�������cp, ��H����S�ֱ����, �Ӷ��õ�Һ�������, �ָ���Cu��Һ�����ݲ����¶ȱ仯��ʵ����ʵ, ���Խ���һ������ֵ��ΪҺ����۵㵽�е�����ݡ�
2 ������
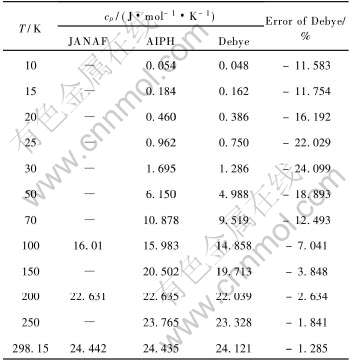
2.1 ��ѹ����
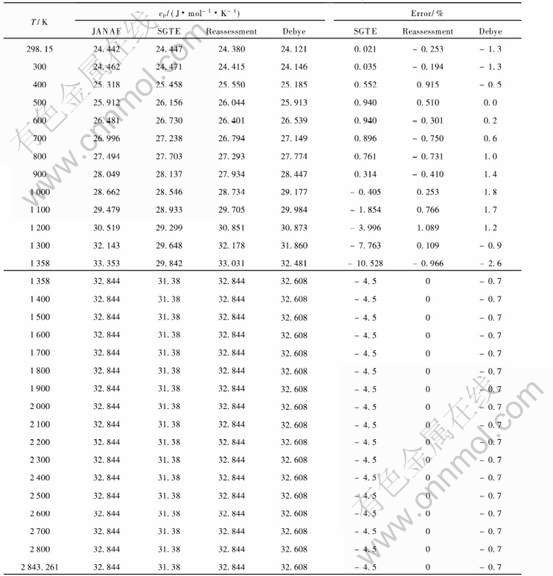
����0~298.15K�¶����������ֱ�Ӳ���Debye-Gr��neisenģ�ͽ��м���, ����ʵ��ֵ���жԱ�(����2)�� ��2�С�AIPH���ǡ�American Institute of Physics Handbook��[10]�ļ�д , ��Debye������Debye-Gr��neisenģ��, ģ�͵ļ��������AIPHΪ���� ����298.15~2843.261K���������, ��SGTE���������ݿ����뱾�������������Ľ����Reassessment����Debyeģ�ͼ��������жԱ�(����3)��
��2 Debye-Gr��neisenģ�ͼ�������Cu ��fcc������º�ѹ������ʵ��ֵ�ĶԱ�
Table 2 Comparison of isobaric heat capacity of fcc phase of Cu metals between experimental data and calculated ones of Debye-Gr��neisen model at low temperure
2.2 ��
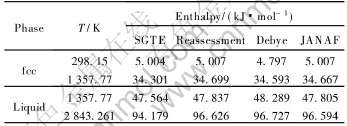
��SGTE���ݿ⡢ ������������� ģ�ͼ���Ľ����JANAF���ݽ��жԱ�(��4��ͼ1)�� ��4����������3�ַ�������298.15K�� �۵�1357.77K�ͷе�2843.261K����ֵ��JANAF���ݵĶԱȡ�
2.3 ��
����ֵ�Ա�����, ��5��ͼ2�������صĽ����
2.4 Gibbs��
������С���˷���SGTE���������ݿ���CuԪ�ص�Gibbs�ܱ���ʽ��������������õ��˱�6���еĽ����
��3 ��SGTE���ݿ⡢ ������������������Debye-Gr��neisenģ�ͼ�������Cu �ĺ�ѹ����ֵ��ʵ�����ݵĶԱ�
Table 3 Comparison of isobaric heat capacity of Cu metals between experimental data and ones of SGTE database, reassessment calculation and Debye-Gr��neisen model
��4 ��ͬ�����о�Cu�Ĺ�Һ��ؼ��¶ȵ����ֵ�Ա�
Table 4 Comparison of enthalpies at key temperatures of Cu by various methods
��5 ��ͬ�����о�Cu�����Һ��ؼ��¶ȵ����ֵ�Ա�
Table 5 Comparison of entropies at key temperatures of Cu by various methods
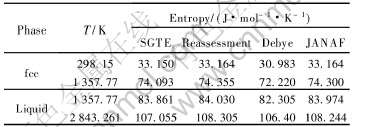
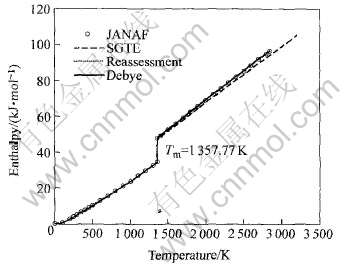
ͼ1 ��ͬ�����о�Cu������ֵ�ĶԱ�
Fig.1 Comparison of enthalpies of Cu by various methods

ͼ2 ��ͬ�����о�Cu��ֵ�ĶԱ�
Fig.2 Comparison of entropies of Cu by various methods
��6 ��SGTE���������ݿ��н���Cu��Gibbs�ܽ�������������Ľ��
Table 6 Reassessed functions of Gibbs energy of SGTE database of pure Cu

��SGTE���ݿ⡢ ��6��������������� ģ�ͼ�������JANAF�����ڱ�7��ͼ3���жԱȡ�
��7 ��ͬ�����о�Cu�����Һ��ؼ��¶ȵ��Gibbs��ֵ�Ա�
Table 7 Comparison of Gibbs energies at key temperatures of Cu by various methods
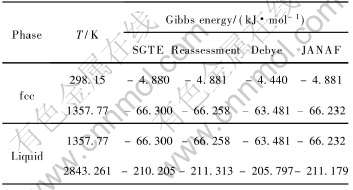
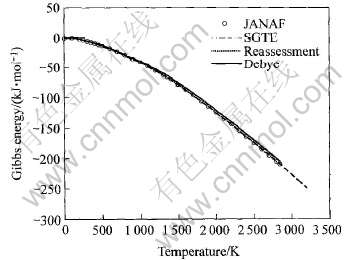
ͼ3 ��ͬ�����о�Cu����Gibbs�ܵĶԱ�
Fig.3 Comparison of Gibbs energies of Cu by various methods
3 ����������
3.1 ����
�ڵ��¶�0~298.15K, ��Debye-Gr��neisen����ģ�ͽ��м���, ͨ����JANAFʵ��ֵ�Լ�AIPH���ݶԱ�, ��������ֵƫС, ��ԭ���������: һ������ģ�ͼ����������Ҫ�Ǿ�������, û�п��ǵ����µ������ݵĹ���[9], ���½��ƫС; ���ǵ°��¶Ȧ�D�����Ǻ㶨�����, ��������������仯[8], ���ڱ������н���D��Ϊ�㶨ֵ������, ���½��ƫС; ����ʵ�ʾ��岢�����뾧��, �и���ȱ�ݴ���, ���Ƕ������нϴ�Ӱ��[19, 20], �����뾧����Ϊģ�������н�������ƫС��
�ڹ�Һת���¶�1357.77K, ���������� ģ�ͼ����SGTE��ת�����ݷֱ�Ϊ: -0.187�� 0.127��1.538J/(mol��K), ��JANAF����-0.509J/(mol��K)���, ����������ƫ����С, SGTE��ƫ�����
��298.15K���ϵĸ��¶�, ��SGTE����ʽ���������Ժ�ļ�������500K����ֱ���е�2843.261K�ĸ������ݺ�ʵ�����ݵ�ƫ���SGTE��ֵҪС; ͬ��, ��400~700K�� 1100K���е�2843.261K, ��Debye-Gr��neisenģ�ͼ���õ��Ľ����SGTE��ȷ��
3.2 ��
���ڵ��¶�0~298.15K����ģ�͵����ݱ�ʵ��ֵƫ��, ���Ա�4��298.15K����ֵΪ4.797kJ/mol, �����������Ķ�Ҫ�͡� ����, ����298.15K���ϵĸ��¶�500��1200K, ����ģ�����ݶ�ʵ��ֵ������ƫ��, �ֲ��˵��µĸ�ƫ� ͬʱ, 400��700K�� 1100���е�2843.261K, ����ģ������ֵ��SGTE��ȷ�� ������ԭ��ʹ�����۵�ʱ, ����ģ�ͼ������õ���ֵ��SGTE�����ݸ���ȷ��
��SGTE����ʽ�������������Ժ�, ���ݼ�������400K���³��ָ�ƫ��, ��500K���ϳ�������ƫ��, �������������, ���������������������Ҫ��SGTE��ȷ, �������۵�ʱ�Ĺ�����ֵ��SGTE���ݿ���ӽ�ʵ��ֵ��
�ڹ�Һת���¶�1357.77K, ���������� ģ�ͼ����SGTE��ת���ʷֱ�Ϊ: 13.138�� 13.696��13.263, ��JANAF����13.138���, ����������ƫ����С, ģ�ͼ���ƫ����� �ɱ�3֪: 3�ַ��������Һ�����ݡ� ģ�ͼ����SGTE���ݾ�Ϊ��ƫ��, ��������Ϊ��ƫ��, ����SGTE��ƫ�����, Ϊ-4.5%, �����е���ֵ����ƫС; ģ�ͼ���ֵ��ƫ���С, Ϊ-0.7%, ���������۵���ֵ���, �����е���ֵƫ��; ��������ֵ���Ч�����, ��е���ֵ��ʵ�����ݷdz��Ǻϡ�
3.3 ��
��298.15K, ģ�ͼ������ֵ����С����������ֵ, ����ֵ��ͬ, ����Ҫԭ�����ڵ�������ֵƫ��; ��Ȼ�������ݳ�������ƫ��, ����ģ�ͼ������ݱ�SGTE����ȷ, ����, �����صĶ��岻ͬ, ������ݵĻ���Ϊ��[SX(]cp[]��[SX)]d��, ���ʵĻ��ַ�ʽ��cpd�Ȳ�ͬ, ��¶�����ͬ���������ݺ���ͬ�Ļ��ּ��㲽��, �����¶ȱ仯����������ҪС, �����¶�Խ��, ��ֵ����ԽС�� ���, ���е��¶�ʱ, ��Ȼģ�ͼ������ֵ��SGTE��, ����ֵ�����ڵ��µ���������ƫС, ������������С, ����һ���¸�ƫ��IJ�����С, ���յ��¼�������SGTE�����С, ��JANAF���ݵ�ƫ�����
���۵�1357.77K, ���������� ģ�ͼ����SGTE��ת���طֱ�Ϊ: 9.675�� 10.085��9.768J/(mol��K), ��JANAF����9.674J/(mol��K)���, ����������ƫ����С, ģ�ͼ����ƫ�����
3.4 Gibbs��
Gibbs�����������صĹ���, ģ�ͼ���ֵ��������ֵ��������������ƫС, �����ض�Gibbs�ܵ�Ӱ����ʸ���, �����С����ֵ�����˽ϸߵ�Gibbs��, ��7��ʾ��ģ�ͼ���ֵ���Ա�����ֵҪ��; ����������SGTE�Ľ����298.15K��1357.77K�dz��ӽ�, ֻ���ڷе��¶�2843.261Kʱ, ͬ����Ϊ��ֵ��������������ƫС, ����Gibbs�ܵ�Ӱ�����ֵ����, �Ӷ�����SGTE��Gibbs�ܸ��ߡ� ��JANAF�������, ���������Ľ��������ϵúܺá�
4 ����
1) ����SGTE���������ݿ���Gibbs�ܵı���ʽ, ���JANAF����ѧʵ������, ����С���˷��Խ���Cu��Gibbs�ܱ���ʽ���������������� ������Debye-Gr��neisenģ�Ͷ�Cu������ѧ���ʽ����˶Ա��о�, ������������ģ�ͼ���Ľ����SGTE��JANAF���ݽ����˶Աȷ����� �������: ���������Ľ���ȷ, �ڹؼ��¶ȵ�, �ر������۵��ת�����ݡ� ת���ʺ�ת���ؾ���SGTE������������JANAFʵ������, ֤����������SGTE���ݿ���Ԫ��Cu��Ȼ̬�����Һ��Gibbs�ܱ���ʽ�����������Ǻ����Ϳɿ���, �������ںϽ������ѧ���㡣
2) ��0~298.15K�ĵ��¶�, Debye-Gr��neisenģ�����ݵļ�������ʵ��ֵƫ��, ��ԭ���������, �������˵��µ������ݵĹ���, �����˵°��¶ȵı仯�����ݵ�Ӱ���Լ������˾���ȱ�ݶ����ݵ�Ӱ��, ��Ϊ����ģ�ͽ�������ָ���˷��� ��298.15K���ϵĸ��¶�, ģ�ͼ���������ڲ����¶�������ʵ�����ݷ��ϽϺ�; ���۵��¶�, ת���ص�ƫ��ϴ�, ����Ҫԭ�����ڼ���ĵ�������ֵƫС��
3) ����Debye-Gr��neisenģ�ͶԹ���Һ��������о�, ͨ�������۵�ʱ���ݡ� �ʺ���ֵ��������, ������Һ������ݵ�����ѧ���ʡ� ��������SGTE���ݿ����ݸ��ӽ�ʵ��ֵ, ֤���Թ���Һ�������ģ���о��dzɹ��ġ�
REFERENCES
[1]Bo S, Fritz A. The Ringberg workshop 1995 on unary data for elements and other end-members of solutions[J]. Reassessmentphad, 1995, 19(4): 433-436.
[2]Saunders N, Miodowik A P, Dinsdale A T. Metastable lattice stabilities for the elements[J]. Calphad-Computer Coupling of Phase Diagrams and Thermochemistry, 1988, 12: 351-374.
[3]Wang Y, Curtarolo S, Jiang C, et al. Ab initio lattice stability in comparison with CALPHAD lattice stability[J]. CALPHAD, 2004, 28: 79-90.
[4]Zhang Z J. Reassessmentculation of the properties of some metals and alloys[J]. Journal of Physics: Condens Matter, 1998,10: 495-499.
[5]Dinsdale A T. SGTE data for pure elements[J].CALPHAD,1991,15(4): 317-425.
[6]Chase M W. NIST-JANAF Thermochemical Tables(Fourth Edition Part I)[M]. Gaithersburg: National Institute of Standards and Technology, 1998.1005-1009.
[7]�sag[DD(-1]��[DD)]ain T. Thermal and mechanical properties of some fcc transition metals[J]. Physical Review B, 1999,59(5): 3468-3473.
[8]LU Xiao-gang. Theoretical Modeling of Molar Volume and Thermal Expansion[D]. Stockholm, Sweden: Royal Institute of Technology, 2005.
[9]Chase M W, Ibrahim A, Alan D, et al. Heat capacity models for crystalline phases from 0K to 6000K[J]. CALPHAD,1995,19(4): 437-447.
[10]American Institute of Physics. American Institute of Physics Handbook, 3rd ed[M]. New York: McGraw-Hill Book Company, 1972: 4-106, 4-119-4-138.
[11]McBride B J, Zehe M J, Sanford G. NASA Glenn Coefficients for Reassessmentculating Thermodynamic Properties of Individual Species[EB/OL]. http://gltrs.grc.nasa.gov/reports/2002/tP-2002-211556.pdf. Glenn Research Center, Cleveland, Ohio, 2002. 9.
[12]Kittel C. Introduction to Solid State Physics, 6th ed[M].New York: John Willey & Sons, Inc, 1986: 55, 57, 144, 110.
[13]Moruzzi V L, Janak J F. Reassessment and calculated thermal properties of metals[J]. Physical Review B,1988, 37(2): 790-799.
[14]Garbulsky G D, Ceder G. Contribution of the vibrational free energy to phase stability in substitutional alloys: methods and trends[J]. Physical Review B, 1996, 53(14): 8993- 9001.
[15]IIDA T, GUTHRIE R I L. The Physical Properties of Liquid Metals[M]. Oxford: Clarendon Press, 1988. 18-32.
[16]Shri S. Liquid Crystals: Fundamentals[M]. Singapore: World Scientific Publishing Co.Pte.Ltd., 2002. 1-23.
[17]XIE You-qing, MA Liu-ying, ZHANG Xiao-dong , et al. Microstructure and properties of Cu-Ni alloys[J]. Science in China(series A), 1993, 36(5): 612-623.
[18]Weast R C. CRC Handbook of Chemistry and Physics, 70th ed[M]. Florida: CRC Press Inc, 1990. B-216.
[19]Ostanin S, Salamatov E. Effect of point defects on heat capacity of yttria-stabilized zirconia[J].Physical Review B, 2003, 68(17):2106-2109.
[20]Wolverton C, Ozolins V, Asta M. Hydrogen in aluminum: first-principles calculations of structure and thermodynamics[J]. Physical Review B, 2004, 69(14): 4109-4124.
(�༭��ѧ��)
������Ŀ: ������Ȼ��ѧ����������Ŀ(50271085, 50471058)
�ո�����: 2005-11-09; ������: 2006-02-23
ͨѶ����: �ջԽ�; �绰: 0731-8879287; E-mail: taohuijin@hotmail.com

