J. Cent. South Univ. (2012) 19: 1796-1801
DOI: 10.1007/s11771-012-1210-3
First-principles investigation of cohesive energy and electronic structure in vanadium phosphides
YANG Zhen-hua(����)1, 2, 3, 4, 5, WANG Xian-you(������)1, 2, 3, 4, 5, SU Xu-ping(����ƽ)5
1. Faculty of Materials, Optoelectronics and Physics, Xiangtan University, Xiangtan 411105, China;
2. Key Laboratory of Low Dimensional Materials and Application Technology of Ministry of Education
(Xiangtan University), Xiangtan 411105, China;
3. School of Chemistry, Xiangtan University, Xiangtan 411105, China;
4. Key Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education
(Xiangtan University), Xiangtan 411105, China;
5. Key Laboratory of Materials Design and Preparation Technology of Hunan Province
(Xiangtan University), Xiangtan 411105, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2012
Abstract: First-principles calculations based on the density-functional theory were employed to study the crystal structure of vanadium phosphide compounds, such as V3P, V2P, VP, VP2 and VP4. Cohesive energy of five types of vanadium phosphide compounds was calculated to assess their structural stability. The charge density distribution and densities of states of vanadium phosphides were discussed to study further their electronic structures. The results show that the structure of metal-rich compounds is considerably more stable than the phosphorus-rich compositions, and covalent bond exists between the V and P atoms of V3P, V2P, VP, VP2 and VP4.
Key words: first-principles calculations; stability; cohesive energy; covalent bond
1 Introduction
In recent years, lithium ion batteries have grown rapidly as new portable power sources. Carbon materials are commercially used as the anode material of lithium ion battery. However, theoretical capacity of carbon materials is only 372 mA��h/g [1]. In order to further enhance energy density of lithium ion batteries, new anode materials need to be developed [2-4].
Transition metal phosphides can provide high theoretically gravimetric and volumetric capacities because they can uptake large amounts of Li through insertion, displacement or conversion reactions. Hence, transition metal phosphides have been studied as a kind of anode materials that could potentially replace graphite in lithium ion batteries [5-9]. Recently, V-P compounds have been aroused great interest due to their high theoretically gravimetric and volumetric capacities. It was reported that VP2 as anode material has reversible capacity of 890 mA��h/g, however, VP2 suffers from bad cyclic stability (irreversible capacity of 37% after 20 cycles) [10]. Many factors can affect cyclic stability of anode material, such as ionic or electronic conductivity and crystal structure stability. It is very essential to explore systematically the nature of V-P compounds as anode material so as to predict anode material with good properties. Although the structures of vanadium phosphides, for example V3P, V2P, VP, VP2 and VP4, have been known [11-15], to our best knowledge, there are less reports about theoretic analysis of transition metal phosphides as anode active material of lithium ion battery. In this work, the stability, bonding mechanism and electron structure of the V-P compounds were investigated by first-principles methods.
2 Models and computational details
Five types of the binary V-P compounds, V3P, V2P, VP, VP2 and VP4, were considered. The crystallographic data of V-P compounds are summarized and listed in Table 1. The sketch of single crystal cell of above compounds is shown in Fig. 1.
The calculations have been performed using the ab-inito total-energy and molecular-dynamics program VASP (Vienna ab-initio simulation program) developed at the Institut F��r Materialphysik of the Universit?t Wien [16-19]. The projector augmented plane-wave pseudopotentials were built within the generalized gradient approximation (GGA) scheme. The exchange- correction energy of Perdew�CBurke�CErnzerhof (PBE) scheme was used for all of the calculations. A kinetic cutoff energy of 400 eV for the plane waves has been performed. Brillouin Zone integrations were performed using Monkhorst and Pack k-point mesh. The k-point grids for V3P, V2P, VP, VP2 and VP4 were 3��3��5, 4��8��3, 9��9��4, 10��10��10 and 10��10��4, respectively. For the calculation of the electronic density of states, the tetrahedron method with Bloch corrections was used with 6��6��10, 6��12��6, 13��13��6, 13��13��13, 14��14��6 for V3P, V2P, VP, VP2 and VP4, respectively. All structure relaxations were preformed with the Quasi Newton (QN) algorithm until the forces on the atoms converged to less than 0.01 eV/?. A spin-restricted approach was used for models of V-P compounds because polarization effects were found to be negligible.
Cohesive energy is an important parameter to assess the stability of V-P alloys, and cohesive energy is often defined as the work which is needed when crystal is decomposed into a single atom. Hence, the more negative the cohesive energy value, the more stable the crystal structure [20]. In order to obtain cohesive energy value correctly, the total energy for the crystal and the free atoms are calculated based on a cubic supercell that contains relative atoms. The cubic supercell has the lattice constant of 15 ?. The cohesive energy of V-P alloys according to the following formula [21-22] is calculated as
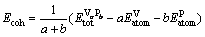
where  is the electronic total energy of primitive cell of VaPb,
is the electronic total energy of primitive cell of VaPb,  and
and are the electronic total energies of the isolated V atom and P atom in freedom states, and a and b refer to the numbers of the V atom and P atom of VaPb units contained in conventional cell, respectively.
are the electronic total energies of the isolated V atom and P atom in freedom states, and a and b refer to the numbers of the V atom and P atom of VaPb units contained in conventional cell, respectively.
Table 1 Crystallographic data of V-P compounds
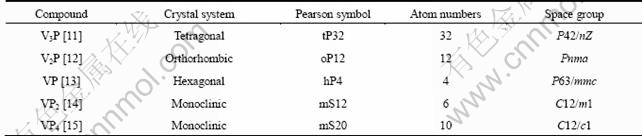

Fig. 1 Crystal structures of V-P compounds: (a) V3P; (b) V2P; (c) VP; (d) VP2; (e) VP4
3 Results and discussion
3.1 Structural properties and stability of crystal
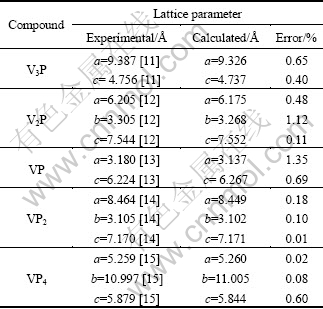
The crystal structures of V-P compounds studied in this work are built based on experimental results. The calculated results and experimental data have both been tabulated in Table 2. It can be seen from Table 2 that for the V-P compounds, the discrepancy is less than 1.5% and the optimized structures are in good agreement with experimental values.
Table 2 Calculated and experimental lattice parameters
It is found through calculation that the cohesive energy of V3P, V2P, VP, VP2 and VP4 are -5.577 8, -5.545 3, -5.265 1, -4.755 7 and -4.234 7 eV/atom, respectively (seen from Fig. 2). Since Ecoh values of all V-P compounds are negative, it implies that there is strong V��P bond between V and P atoms. Besides, it can be concluded from Fig. 2 that the metal-rich compounds are considerably more stable than the phosphorus-rich compositions.
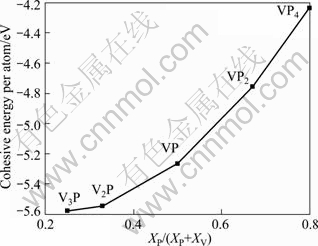
Fig. 2 Calculated zero-temperature cohesive energy of V-P compounds
3.2 Electronic structure and bonding mechanism in V-P compounds
In order to insight into the bonding mechanism in V-P compounds, the total density of states (TDOS) and the partial density of states (PDOS) of V3P, V2P, VP, VP2 and VP4 primitive cells are calculated.
The density of states in V-P compounds are shown in Fig. 3. Zero of x-axis corresponds to Fermi level. Top panels are for TDOS and the two lower panels are for PDOS. It can be found from the Fermi level that V-P compounds are all metallic characters. For V-P compounds, V3P compound is considered as a typical example. The valence band can be divided into two energy areas according to the PDOS. One is from -15 eV to -10 eV, which mainly has a strong P 3s character; the other is from -7.5 eV to Fermi level, which mainly has V 3d character and P 3p character. The valence band located between -7.5 eV and Fermi level is because a strong bonding possesses the characteristic of significant p-d hybridization between V 3d and P 3p states. Analysis of the total density of states (DOS) for V3P, V2P, VP, VP2 and VP4 was performed in the energy range between -7.5 eV and 0 eV.
Figure 4 shows the bonding electron numbers per atom of V-P compounds. It is evident from the above results that the numbers of bonding electron shift to lower value with increasing P element in crystal. When bonding electron numbers decrease, the interaction between valence electron of crystal is impaired, so the stability of crystal structure will become worse. Hence, V3P phase has the highest structural stability, while VP4 phase has the lowest structural stability.
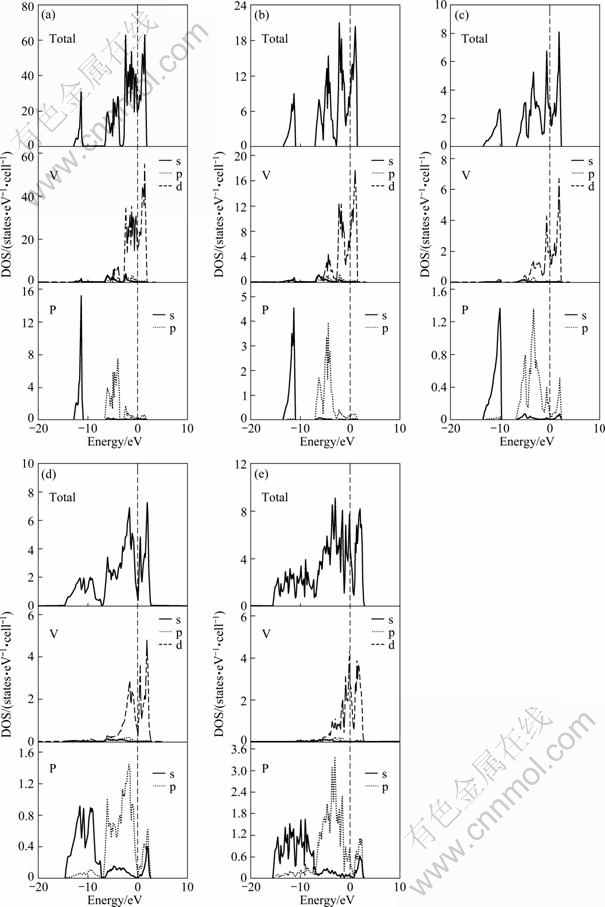
Fig. 3 Total and partial DOS of V-P compounds with different type structures: (a) V3P; (b) V2P; (c) VP; (d) VP2; (e) VP4
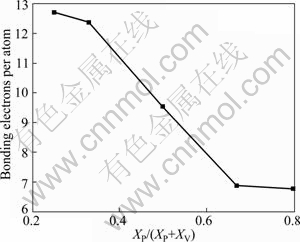
Fig. 4 Calculated bonding electrons per atom of V-P compounds
To further discuss the bonding nature in V-P compounds, the valence electron density of distribution was plotted and analyzed.
The (010) or (001) planes with the valence electron density distribution for V-P compounds are shown in Fig. 5. The remarkable charge distribution overlap between V and P atoms is observed. Apparently, the feature of covalent bond emerges between V and P atoms of V3P, V2P, VP, VP2 and VP4. The charge density overlapping between V and P atoms is reduced with the increase of P element content. This result is in a good agreement with the density of states (DOS) and bonding electron number discussed above.

Fig. 5 Contour plot of total valence charge density on (001) plane for V3P (a), (010) plane for V2P (b), (010) plane for VP (c), (010) plane for VP2 (d) and (001) plane for VP4 (e)
4 Conclusions
1) Metal-rich compounds are considerably more stable than the phosphorus-rich compositions, and the V3P compound has the highest structural stability. The decrease of the structural stability of V-P compounds with the increase of P element is attributed to a decrease in the bonding electron numbers at lower energy level below Fermi level.
2) The electronic structures of V-P compounds (V3P, V2P, VP, VP2 and VP4) investigated with density functional theory show metallic character.
3) Covalent bond exists between the V and P atoms of V3P, V2P, VP, VP2 and VP4.
References
[1] LI Qing-yu, HU Si-jiang, WANG Hong-qiang, WANG Fang-ping, ZHONG Xin-xian, WANG Xin-yu. Study of copper foam-supported Sn thin ?lm as a high-capacity anode for lithium-ion batteries [J]. Electrochimica Acta, 2009, 54: 5884-5888.
[2] ZOU Lin, GAN Lin, KANG Fei-yu, WANG Ming-xi, SHEN Wan-ci, HUANG Zheng-hong. Sn/C non-woven ?lm prepared by electrospinning as anode materials for lithium ion batteries [J]. Journal of Power Sources, 2010, 195: 1216-1220.
[3] WANG Fei, ZHAO Ming-shu, SONG Xiao-ping. The improved electrochemical performance of SnSb-based alloy anode materials for Li-ion batteries [J]. Journal of Alloys and Compounds, 2009, 472: 55-58.
[4] LIU Ping, ZHANG Zhi-an, LI Jie, LAI Yan-qing. Effects of carbon sources on electrochemical performance of Li4Ti5O12 composite anode materials [J]. Journal of Central South University of Technology, 2010,17(6): 1207-1210.
[5] HAYASHI A, INOUE A, TATSUMISAGO M. Electrochemical performance of NiP2 negative electrodes in all-solid-state lithium secondary batteries [J]. Journal of Power Sources, 2009, 189: 669-671.
[6] MAUVERNAY B, DOUBLET M L, MONCONDUIT L. Redox mechanism in the binary transition metal phosphide Cu3P [J]. Journal of Physics and Chemistry of Solids, 2006, 67: 1252-1257.
[7] BOYANOV S, BERNARDI J, BEKAERT E, M?N?TRIER M,DOUBLET M L, MONCONDUIT L. P-Redox mechanism at the origin of the high lithium storage in NiP2-Based batteries [J]. Chemistry of Materials, 2009, 21: 298-308.
[8] PARK C M, SOHN H J. Tetragonal zinc diphosphide and its nanocomposite as an anode for lithium secondary batteries [J]. Chemistry of Materials, 2008, 20: 6319-6324.
[9] XIANG J Y, WANG X L, XIA X H, ZHONG J, TU J P. Fabrication of highly ordered porous nickel phosphide ?lm and its electrochemical performances toward lithium storage [J]. Journal of Alloys and Compounds, 2011, 509: 157-160.
[10] GILLOT F, M?N?TRIER M, BEKAERT E, DUPONT L, MORCRETTE M, MONCONDUIT L, TARASCON J M. Vanadium diphosphides as negative electrodes for secondary Li-ion batteries [J]. Journal of Power Sources, 2007, 172: 877-885.
[11] JAWAD H, LUNDSTROEM T, RUNDQVIST S. Refinement of the crystal structure of V3P [J]. Physica Scripta, 1971, 3: 43-44.
[12] BERGER R, TERGENIUS L E. The crystal structure of V2P [J]. Acta Chemica Scandinavica: Series A, 1976, 30: 387-389.
[13] FJELLVAG H, KJEKSHUS A. Structural properties of VP1-xAsx; x=0-1 [J]. Acta Chemica Scandinavica, 1986, 117: 773-781.
[14] GOELIN M, CARLSSON B, RUNDQVIST S. Refinement and crystal structure of VP2 [J]. Acta Chemica Scandinavica: Series A, 1975, 29: 706-708.
[15] JEITSCHKO W, FLOERKE U, SCHOLZ U D. Ambient pressure synthesis, properties, and structure refinements of VP4 and CoP2 [J]. Journal of Solid State Chemistry, 1984, 52: 320-326.
[16] KRESSE G, HAFNER J. Ab initio molecular-dynamics simulation of the liquid-metal�Camorphous-semiconductor transition in germanium [J]. Physical Review B: Condensed Matter, 1994, 49: 14251-14269.
[17] KRESSE G, FURETHM?LLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Computational Materials Science, 1996, 6: 15-50.
[18] KRESSE G, FURETHM?LLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Physical Review B: Condensed Matter, 1996, 54(16): 11169-11186.
[19] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Physical Review B: Condensed Matter, 1999, 59(3): 1758-1775.
[20] ZHOU D W, LIU J S, PENG P, CHEN L, HU Y J. A first-principles study on the structural stability of Al2Ca, Al4Ca and Mg2Ca phases [J]. Materials Letters, 2008, 62: 206-210.
[21] DENG Yong-he, WANG Tao-fen, ZHANG Wei-bing, TANG Bi-yu. Crystal structure of Mg3Pd from first-principles calculations [J]. Transactions of Nonferrous Metals Society of China, 2008, 18: 416-420.
[22] WANG X F, Li W, FANG G P, WU C W, LIN J G. First-principles calculations on the electronic structure and cohesive properties of titanium stannides [J] Intermetallics, 2009, 17: 768-773.
(Edited by YANG Bing)
Foundation item: Project(20871101) supported by the National Natural Science Foundation of China; Project(09C945) supported by the Scientific Research Fund of Hunan Provincial Education Department, China
Received date: 2011-04-20; Accepted date: 2011-08-25
Corresponding author: WANG Xian-you, Professor, PhD; Tel: +86-731-58292060; Fax: +86-731-58292061; E-mail: wxianyou@yahoo.com

