DOI: 10.11817/j.issn.1672-7207.2016.10.006
���-��-D-���ľ���յĺϳɼ�����
���ȣ����������������⣬����٣��¹��£�Ҷѩ
(��̶��ѧ ��ѧѧԺ�������Ѻû�ѧ��Ӧ��ʡ�������������ص�ʵ���ң����� ��̶��411105)
ժҪ���������������ǰ���������D-ľ��Ϊԭ�ϣ���ȫ��������C1λѡ��������������ת�������������ǰ�������һϵ�д�Ϊ������֮����ż����Ӧ���ѱ�����ѡ���Եغϳ�9�ֲ�̼ͬ�����ȵ����-��-D-���ľ���ա�Ŀ�껯���ᆳ�˴Ź������Խṹ���б���������ƫ���������ȷ����ǵȶ������ܽ��в��ԡ��о���������������������ɻ�-��-D-���ľ�����ڵ�Ũ��ʱ��ʹ���������½����ϵ�ֵ���Ҿ��нϺõķ����ԣ����ɻ�-��-D-���ľ�����нϺõ��黯�ԣ����������n��9ʱ�����-��-D-���ľ���յ��ܽ��ض����¶����߳��½����ƣ���nΪ7��8ʱ�������վ��нϴ���ܽ��ʣ�Ŀ�껯���������Һ������
�ؼ��ʣ�ľ�ǣ����-��-D-���ľ���գ����ݣ��黯����������
��ͼ����ţ�O629.11+3 ���ױ�־�룺A ���±�ţ�1672-7207(2016)10-3323-09
Synthesis and properties of alkyl ��-D-xylopyranosides
KUANG Na, WU Guilong, CHEN Langqiu, XIA Shu, LI Zhencao, CHEN Guoyong, YE Xue
(Key Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education,
College of Chemistry, Xiangtan University, Xiangtan 411105, China)
Abstract: Using the trichloroacetimidate method and taking D-xylose as a raw material, nine kinds of carbon chain length alkyl ��-D-xylopyranosides were synthesized selectively through acetylation, selective deacetylation at C1 position, and conversion to trichloroacetimidate, coupling with a series of acceptors alcohols and deprotection. The structures of target compounds were characterized by NMR technology, and their properties were tested by polarization microscopy(POM), thermal gravimetric analysis (TGA), etc. The results show that the n-octyl and n-nonyl ��-D-xylopyranosides are able to make surface tension decrease to a low value and possess better foaming properties, while n-nonyl ��-D-xylopyranoside has better emulsification. However, with the increase of temperature, dissolution entropies of all alkyl ��-D-xylopyranosides decrease when the chain length of alkyl group is less than 9. The dissolution enthalpies of D-xylopyranosides are bigger when the chain length of alkyl groups are 7 and 8. All alkyl ��-D-xylopyranosides can form thermotropic liquid.
Key words: xylose; alkyl ��-D-xylopyranoside; surface tension; foaming; emulsification
������P�������ﲻ��Ϊ�������������ṩ��Ҫ������[1]�������ں��ᡢ�����ʡ�������֬���Ĵ������������е�λ���أ�������ϵ���ᡢ�����ʺ�֬��������ӵ���Ŧ���á����������Ϊ���������нṹ�������ˮ�Խṹ���飬�뵰���ʺ�֬�����Ƚ�ϳ��ǵ�����֬��Ӱ�������ǵ��۵�������Ӧ[2-3]��ľ���DZ���ֲ��ϸ���ڳɷ��к����ӵ�2����ľ����(xylan)��ʽ���ڵ���ḻ����[4]��ľ���������Ӷ��ǣ���1,4-��-D-ľ�Dzл�������������������Ц�-L-�������ǵȣ���ũҵ�������������ᡢ���ѿǵ������������ϸ�[5]������ͨ����ⷨ��Ч������Ӧ��ľ�ǡ�����������ۼ��ḻ���ǻ��ڶ࣬�����������ˮ�ԣ�ͨ���ṹ���죬���仹ԭ����֬�����Ĵ��������γɸ߱�����Ե�˫������O-���շ��ӣ����ϱ�����Լ��ĽṹҪ�Ӷ��ɿ����������������Լ�[6-7]�����������������õ���Ҫԭ��Ϊ��������֬�����Ĵ������п������ԣ��ڽṹ����ѧ��ҽѧ��ϴ�Ӽ���������������й�����Ӧ�ü�ֵ���������վ�����Դ�ḻ����̬�����Ժá����������á����ԡ��ȶ�ø��ˮ��Һ�еĴ��������ã����Ͽɳ�����չҪ��Ϊ���͵ı�����Լ�[6-9]�����-��-D-���ľ���տɲ���ø������ѧ�ϳɷ��ϳɡ�����ø����ѡ���Ժá����ʸߡ��Ʊ������ºͣ�������ø��ɸѡ�����ԵIJ���ܼ���ɸѡ��ø�ٷ�Ӧ��ƽ����ʱ���ؼ����ӵķ��봿��������[9-13]������һ����������Ϊ����ʹD-ľ�ǻ�ľ�����봼����Fischer���ջ���Ӧ�����ڲ����¶Ƚϸߺͷ�Ӧʱ��ϳ������ﹹ�ͷǵ�һ���������ѵ����⣻ת���շ��õ���ҲΪ�������ѵĻ�������գ�����Һ���������Ȼʹ��ӦЧ����ߣ�����������Һ����Ⱦ��Ʒ������[14]��Koenigs-Knorr��Ϊһ�־��������ѡ���Ժϳ����յķ��������ľ���ȶ��Խϲ�ر����ֽ⣬����Ҫ�������κ��ε��ؽ���������Ϊż����Ӧ�Ĵ�������������ؽ�����Һ�ۻ���������Ⱦ�����������ڷ�Ӧ����Ч���뱣�����ŵ�ѡ��������[9, 15]����ȻPETROVI ��[16]����ά����B12Ϊż����Ӧ�Ĵ����������ڳɱ����ߵ����⡣����ȫ��������ľ��(2)�����Ȼ���������һϵ��֬��������ż����Ӧ����ʱ��Ҫ�ϸ���ƣ������в���ת���ɦ�-�칹�壬��ʱ�����ʱ�������γ��Ԧ�-����Ϊ���IJ��������2-λ�ѱ��������ո�����[17]���ο����-��-D-�������յȺϳ��������о��ɹ�[8-9,15,18-21]������Scheme1��ʾ��·�ߣ���D-ľ��(1)Ϊԭ��ͨ������������1-λ�����������������淴Ӧ���õ���Ӧ�����������ǰ���(4)���������ڹ���1,2-��ʽ���ռ������������ǰ��������������������������ջ�ż����Ӧ�Ĵ�����ʹ�þ����ڻ�����ЧӦ�����������ǰ���(4)����Ӧ��֬������Ӧ����Ӧ�����������������ϳ�һϵ�����-��-D-���ľ���գ���ͼ1��ʾ���������߶����ܽ��ԡ������������黯���ܡ�Һ�����ܡ����ȶ��Ե��������ʽ����о���
��[16]����ά����B12Ϊż����Ӧ�Ĵ����������ڳɱ����ߵ����⡣����ȫ��������ľ��(2)�����Ȼ���������һϵ��֬��������ż����Ӧ����ʱ��Ҫ�ϸ���ƣ������в���ת���ɦ�-�칹�壬��ʱ�����ʱ�������γ��Ԧ�-����Ϊ���IJ��������2-λ�ѱ��������ո�����[17]���ο����-��-D-�������յȺϳ��������о��ɹ�[8-9,15,18-21]������Scheme1��ʾ��·�ߣ���D-ľ��(1)Ϊԭ��ͨ������������1-λ�����������������淴Ӧ���õ���Ӧ�����������ǰ���(4)���������ڹ���1,2-��ʽ���ռ������������ǰ��������������������������ջ�ż����Ӧ�Ĵ�����ʹ�þ����ڻ�����ЧӦ�����������ǰ���(4)����Ӧ��֬������Ӧ����Ӧ�����������������ϳ�һϵ�����-��-D-���ľ���գ���ͼ1��ʾ���������߶����ܽ��ԡ������������黯���ܡ�Һ�����ܡ����ȶ��Ե��������ʽ����о���
1 ʵ��
1.1 �Լ�������
����Ϊ��DM-LM-P��ƫ������(�¹�leiea��˾)��BRUKER-AVANCE-400�ͺ˴Ź�����(��ʿBruker��˾)��X-4������ʾ���۵�ⶨ��(���Ϲ�����Ӣ��������)��TGAQ50���ȷ�����(����PE��˾)��DP-A��������ѹ���¶ȼ�(�Ͼ�ɣ�������豸��)��TLC����ʹ���ൺ�������ֳ���Ϳ����Ϊ0.20~0.25 mm��HF254�轺�塣
�Լ�Ϊ����ɫ�۲��������Ϊ30%������״���Һ��ʹ���ൺ��������0.075~0.150 ��m�轺������ɫ���롣�����Լ���Ϊ���ۻ�ѧ�����������ʵ����ˮ��Ϊ����ˮ��
1.2 ������-��-D-���ľ����(6e)�ĺϳ�
��100 mL��Բ����ƿ�����μ���2.20 g (5.23 mmol)��ѧ��4(��ͼ1)[22]��10 mL���ȼ���(4��10-10 m����ɸ����)��2.48 mL(15.69 mmol)�������������±�ˮԡ��ȴ��0 �����ң��μ�0.65 mL (5.23 mmol)��������������Һ�����跴Ӧ5 h��TLC(ʯ������������������ȼ�V(ʯ����):V(��������)=2:1)��ⷴӦ��ȫ����ӦҺ���ξ�����̼������ˮ��Һ�ͱ���ʳ��ˮ��Һϴ�ӣ�������ˮ�����Ƹ�����ˣ���ҺŨ����������(V(ʯ����)��V(��������)=10:1)���룬�õ���ɫ���廯����5e(1.12 g������55%)��������һ���ѱ�����Ӧ��
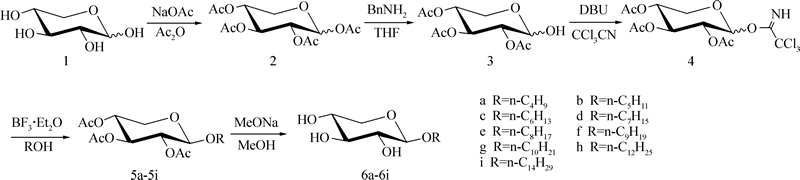
ͼ1 6a~6iĿ�껯����ĺϳ�·��
Fig. 1 Synthetic route toward target compounds 6a~6i
��100 mL��Բ����ƿ�м���5.00 g (12.87 mmol)������5e��40 mL��ˮ�״��������ܽ⣬�ü״��Ƶļ״�ϡ��Һ(�״���������״�ϡ���Ϊ1:5 g/mL)������Һ��pH��Լ10.0�����·�Ӧ5 h��TLC(��������)��ⷴӦ��ȫ����732����������֬���ڷ�ӦҺ��pH=7�����ˣ���ҺŨ����������(��������)���룬�õ���ɫ���廯����6e(2.74 g������81%)��
�������-��-D-���ľ����(������6a~6d��6f~6i)�ĺϳɣ��ֱ�����Ӧ�Ĵ����棬�ϳɷ���ͬ�ϡ�
1.3 �ܽ����ܲ���
�ο�����[8]����25 ��ⶨĿ�����6a~6f�ڲ�ͬ�ܼ��е��ܽ��ԡ�����ⶨ�������£�1) �ڵ�����ƽ��ȷ��ȡ������Ŀ����������Ա��У�2) �ʵ�����Ԥ�����ܼ�������ҡ����������ҡ1.0 h��ʹ�����ܼ��дﵽ�ܽ�ƽ�⣻3) �������ܽ��������������ȷ�������ܼ�������ҡ���ϼ�����ҡ����δ�ܽ�Ľ��٣����Ϊ�μӣ�4) �۲��ܽ�����������������ʿ�����Һ��ʱ������Ϊȫ���ܽ⣻5) ���������ܼ�������������õ����ڸ��¶��µ��ܽ�ȡ�
1.4 �ܽ��ʵIJⶨ
�ο�����[21-22] ���ֱ���������6a~6f��15��25��35��45��55 ��ʱ�ܽ��S3���ٸ���ʽ(1)��������ľ���յ��ܽ���(��solH)��
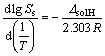 (1)
(1)
��Gibbs-Duhem��ʽ�Ƶ�����
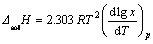 (2)
(2)
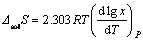 (3)
(3)
���ܽ���(��solS)Ϊ
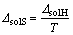 (4)
(4)
1.5 �黯���ܲ���
�ο�����[8]������Һ�ֱܷ�ȷ��ȡ20 mL��������Ϊ0.12%�����-��-D-���ľ����ˮ��Һ��20 mL����100 mL������Ͳ�У��Ǻ�ƿ������ֻ�Ͼ��Ⱥ���1.0 h���۲��¼��Һ������(Veb)��ˮ�����(Vwb)����ͬ���ķ����ⶨ���-��-D-���ľ���նԲ����͵��黯���ܣ��۲��¼��Һ�����(Vez)��ˮ�����(Vwz)��
1.6 ����������ĭ���ȶ��Բⶨ
�ο�����[8]��������������Ϊ0.12%�����-��-D-���ľ����ˮ��Һ100 mL������Һ��ȷ��ȡ10.0 mL��100 mL������Ͳ�У��Ǻ�ƿ����Ȼ�����¾�����1 min������������ĭ�ĸ߶�H0��5 min֮���ٴβ�����ĭ�ĸ߶�H5������ʽ������ĭ��ʧ�ٶȦԣ�����������ĭ���ȶ��ԣ�
��=(H0-H5)/(60��5) (5)
��Ȼ��H0Խ����������Խǿ����ԽС������ĭ����ʧ�ٶ�Խ������ĭ���ȶ���Խǿ��
1.7 ���������IJⶨ
�ο�����[23]�����������ѹ��������һϵ�в�ͬ�������������-��-D-���ľ����(6c~6f)ˮ��Һ��25 mL���ֱ�������25 ��ʱ�����ѹ����Ȼ����㲻ͬ���������µ����ľ���յı���������
1.8 ���ȶ��Բ���
��TGA(�ȷ�����)�ϲ������-��-D-���ľ���յ����ȶ��ԡ���N2��Ϊ�������壬��20 ��/min���ٶ����£���������õ���������ʧ���߷����ж��ȷֽ��¶ȡ�
1.9 ����Һ���Թ۲�
����DM-LM-P��ƫ����������3 ��/min���������ʹ۲����-��-D-���ľ���յ�����Һ�����ԡ�
2 ���������
2.1 ���-��-D-���ľ���պϳɷ�����1H NMR���������乹��ȷ��
�ڲο�����[16��18]�Ļ����ϣ�ֱ�Ӳ���ȫ��������ľ��(2)�������������Ѵ�����һϵ��֬��������ż����Ӧ��TLC���ٷ�Ӧ���̣����ֳ�������ֽ�Ŀ��㣬���봿������̫���ʽϵͣ�ʵ��Ӧ�ü�ֵ���ߡ���ͼ1��ʾ���·�ߣ�ͨ��������ż�����ѱ������ǻ�ѧ���ԣ���Ч��ɸ������-��-D-���ľ����(6a~6i)�ĺϳɡ�
���ľ�ǻ����������ǰ���(4)�봼����ż����Ӧ�õ����-2,3,4-��-O-������-��-D-���ľ����(5a~5i)������ͨ���������������������Ӧ�ĸ������-��-D-���ľ����(6a~6i)�����۵㡢1H NMR�������1���ӱ�1�ɼ����������-��-D-���ľ����1H NMR��H-1��ѧλ�ƺ���ϳ����ֱ�Ϊ6a(�� 4.40 (d, J1,2 = 5.3 Hz))��6b(�� 4.29 (d, J1,2 = 7.7 Hz))��6c(�� 4.31 (d, J1,2 = 6.5 Hz))��6d(�� 4.29 (d, J1,2 = 6.8 Hz))��6e(�� 4.33 (d, J1,2 = 6.2 Hz))��6f(�� 4.34 (d, J1,2 = 6.3 Hz))��6g(�� 4.36 (d, J1,2 = 5.9 Hz))��6h(�� 4.34 (d, J1,2 = 6.2 Hz))��6i(�� 4.07 (d, J1,2 = 7.4 Hz))����1H NMR��H-1��ѧλ�ơ�����4.07~4.40֮�䣬��ϳ���J1,2��5.3~7.7 Hz��Χ�ڣ�˵�����ϳɵ����-��-D-���ľ����6a~6i�����ռ���Ϊ1,2��ʽ�Ħ�-���ռ�[16-17]��
��1 ���-��-D-���ľ���յ��۵��1H NMR���
Table 1 Results of melting point and 1H NMR of alkyl ��-D-xylopyranoside

2.2 ���-��-D-���ľ���յ��ܽ���
�����������£��ϳɵ����-��-D-���ľ����(6a~6i)��ˮ���״����Ҵ������������е��ܽ��(S1)��ͼ2��ʾ����ͼ2�ɼ����������̼��n��6ʱ��ľ������ˮ�е��ܽ�Ƚϵͣ��������̼��n��10ʱ������������ˮ���ڼ״����Ҵ��У�ľ��������������������ܽ�����½����������������У�ľ���յ��ܽ�Ⱦ��ϵ͡�
Ϊ�ˣ���һ���ⶨ���-��-D-���ľ����(6a��6c~6h)�ڸ��ִ�(�״����Ҵ����������������������촼��������)���ܽ��(S2)�������ͼ3����ͼ3�ɼ��������������£��������������������ľ������ͬһ�ִ��е��ܽ�����½�����ͬһ�����ľ���յ��ܽ��Ҳ���Ŵ���̼�����������½������ԣ����������Ҵ��ȶ�����(C1~C6)��ˮ��ͬ���������ľ���յ��ܽ�ȡ�

ͼ2 ľ������ˮ���״����Ҵ������������е��ܽ���
Fig. 2 Solubility of xylopyranoside in water, methanol, ethanol and ethyl acetate

ͼ3 ľ������ˮ�Ͳ�ͬ���е��ܽ���
Fig. 3 Solubility of xylopyranoside in water and various alcohols
2.3 ���-��-D-���ľ���յ��ܽ���
��ͬһ�¶��£����-��-D-���ľ�������ž�����ˮ���õ���������ȵ���������ˮ������������Ϊ��ˮ�е��ܽ����С���ڲ�ͬ�¶��£������¶����ߣ�ͬһ���ľ�����ܽ������Ϊ�˲ⶨ���ľ���յ��ܽ��ʺ��ܽ��أ��ֱ�ѡȡ��������ˮ����һ���ܽ�ȵ����-��-D-���ľ����6a~6f�����15��25��35��45��55 ���µ��ܽ�ȣ�����ʽ(1)��ʾ���¶Ⱥ��ܽ�ȹ�ϵʽ(����TΪ����ѧ�¶ȣ�RΪĦ�����峣������solHΪ�ܽ���)����ͼ4�ɵõ���ͬ�¶����ܽ������б��(-��solH/(2.30R))���Ӷ���������-��-D-���ľ����6a~6f���ܽ���(��2)������ʽ(4)�����Ӧ���ܽ���(��solS)���¶ȵĹ�ϵ���ߣ���ͼ5��ʾ��
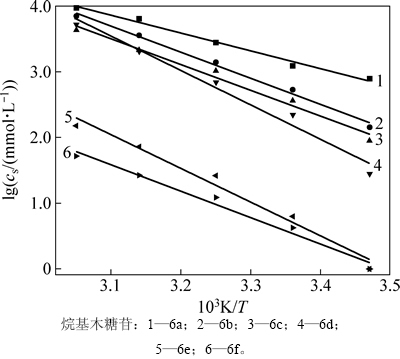
ͼ4 ľ�����ڲ�ͬ�¶��µ��ܽ���
Fig. 4 Solubility of xylopyranoside atvarious temperature
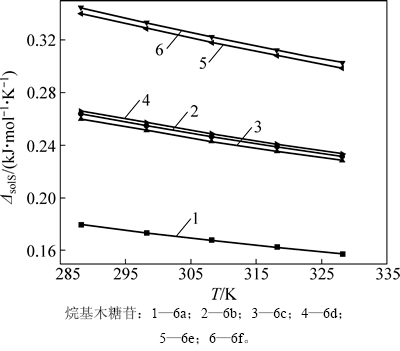
ͼ5 ľ���յ��ܽ��ء�solS
Fig. 5 Entropy of xylopyranoside
��2 ľ����6a~6f���ܽ���
Table 2 Solution enthalpy of xylopyranoside 6a~6f

�ܽ���Ϊ��ֵ��ζ����������ˮ���ܽ������һ�����ܹ��̡�ʵ���ϣ��ܽ���һ�������ӵĹ��̣�����������ˮʱ�ƻ��ܼ�ˮ������ϵ���еĴ���ʹˮ����֮��ǿ�����������������������ˮ����֮�����������ڵĸ�������������������ʹ���ܽ��������ų��������������ֲ��ܼ�ˮ����֮����������������Ҫ�����������ԣ��¶�������ṩ�������������������ܼ�ˮ�е��ܽ�������ǿ����ͼ5�ɼ�����������ˮ���ܽ�ʱ���ܽ��س������¶��������½������Թ�ϵ�����ж���-��-D-���ľ����6a���ܽ�����ͣ���̼����4��(������6a)������7����8��������-��-D-���ľ����6d������-��-D-���ľ����6eʱ�ܽ��شﵽ�����������½�(������6f)��
2.4 ���-��-D-���ľ���յ��黯��
���-��-D-���ľ���յı�����������������ij����йء�ͼ6��ʾΪ������6a~6f�Բ����ͺͱ����黯���ܡ���ͼ6���Կ�����������6a~6f�Ա��Ͳ������黯����1 h��������ˮ�����(Vwb��Vwz)����������������������٣�����������ľ����6a~6f�Ա��Ͳ����͵��黯�����������������������ǿ����n=9ʱ(������6f)���Ա��Ͳ����͵��黯�������ﵽ������������ɻ�-��-D-���ľ����(6f)��������ϳ�������ˮ���л����������������ǿ���Ӷ�ʹ�����黯����(���Ͳ�����)�����γɽ���Ĥ��ǿ����Ӧ���ӣ���״ҺҺ��۽�ʱ�ܵ����������γ���״Һ���ȶ�����ߣ��Ӷ��ܹ��γɸ����ȶ�����Һ�㡣

ͼ6 ľ���յ��黯��
Fig. 6 Emulsifying property of xylopyranoside (6a~6f)
2.5 ���-��-D-���ľ���յ�����������ĭ�ȶ���
ľ����(6a��6f)���������ܼ�ͼ7��ͼ7��������ľ���յ��������n��6(������6c~6f)ʱ���������õ������ԣ�����������������ӣ�������Ҳ����ǿ����n=8ʱ����������ã�����ּ�С����n=9ʱ���ɻ�-��-D-���ľ����(6f)�ڵ�����������(0.12%)���к�ǿ�ķ�����������ĭϸ�塣��ĭ�ȶ���(��)��������������ӳ��ֳ����½������������ƣ���nΪ8��9ʱ��ǿ����Ϊ�������ɻ�ľ����(n=8��9)����ˮ�Ի�������ˮ�Ի����ܹ����õ�ƥ�䣬��Һ������γɵ�ҺĤǿ�ȱȽϴ�����������ǿ��
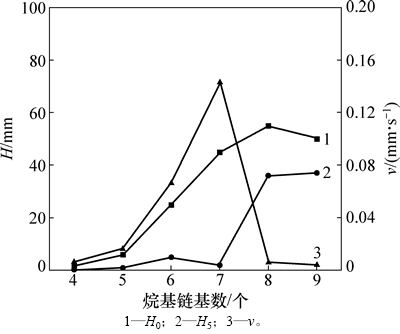
ͼ7 ľ����(6a~6f)����������
Fig. 7 Foaming capability of xylopyranoside (6a~6f)

ͼ8 ľ����(6c~6f)�ڲ�ͬ����Ũ���µı���������
Fig. 8 Surface tension of xylopyranoside (6c~6f) in different concentrations
2.6 ���-��-D-���ľ���յı�������
���-��-D-���ľ���յı�����Կ���������Һ���ͱ���������������Ч����������ǰ���ñ�����Լ�ʹ�ܼ������������ͳ̶���������������ʹ������������һ��ֵʱ����Ҫ�ı�����Լ�����������������ͨ�������黯���ܺ��������ܲ��Խ�����Կ�����ľ����6a,6b�ı�����Խϲ����6c~6f�������õı�����ԡ����IJ��������ѹ������25 ���£��ⶨ���������ͬ�������������-��-D-���ľ����6c~6f����Ӧ�ı�����������ͼ8���Եó���1) �������-��-D-���ľ����(6c~6f)���ӵ�ˮ�����Եؽ�����ˮ�ı������������ֳ����ṹ������ľ���շ��ӹ��еı�����ԣ�2) �����������������Χ�ڣ����ľ����(6c~6f)���������������������������ȼ����½������ֻ����½�����������������ƣ�3) ��ͬ��ľ����6c~6f�ٽ���������(CMC)��ͬ��
��3��ʾΪ���-��-D-���ľ����6c~6f���ٽ���������������Ӧ�ı�����������Һ������������ﵽ����ʱ(�պñ���ʱ��Ũ�ȼ�Ϊ�ٽ�Ũ��)������������С���ӱ�3���Կ���������-��-D-���ľ����(6e)�ﵽ�ٽ�Ũ��ʱ��Ӧ�ı�����������͵ģ�������ǿ����Ϊ���ľ���յı��������������ˮ�ǻ�����ˮ�������ͬ���������������Ϊn=8(����6e)ʱ����ˮ�Ժ���ˮ�Դﵽ���ƽ�⣬����������͡�
��4��ʾΪ����6c~6f��ͬһŨ��ʱ��Ӧ�ı����������ӱ�4�ɼ���������6c~6f����Ũ�ȶ�Ϊ0.3 g/Lʱ������������������������������С������6fʹ���������½���ͣ�Ч����ߡ���ԭ����������յ������Խ������ˮ��Խǿ����������������Һ�ڲ�����������Һ�����������������������Ա��ֳ��������̼�����ȵ����Ӷ���������ơ�
2.7 ���-��-D-���ľ���յ����ȶ���
ͼ9��ʾΪ��������ʧ�������ͼ9�ɼ������-��-D-���ľ����6a~6i ��ֻ��1��������ʧ̨�ף���ʼ�ֽ��¶ȷֱ�Ϊ157.9��177.3��172.6��162.3��164.1��169.5��151.7��170.4��208.9 �����ң����ֽ�����ʱ���¶�����Ϊ283.2��287.8��296.6��257.9��290.4��286.1��301.9��262.7��339.8 �档����6a~6i �ȷֽ���ʱ���¶ȷֱ�Ϊ303.1��298.1��329.6��365.5��320.4��340.2��332.0��374.6��356.2 �档��������ʧ��Ϊ94.9%��89.9%��91.0%��91.9%��95.3%��96.6%��89.6%��88.5%��91.5%������������ϳɵ����-��-D-���ľ������150 ���������ȶ��ġ�
��3 ���-��-D-���ľ����6c~6f���ٽ罺���������������Ӧ�ı�������
Table 3 Critical micelle concentration (CMC) and surface tension of alkyl ��-D-xylopyranoside 6c~6f
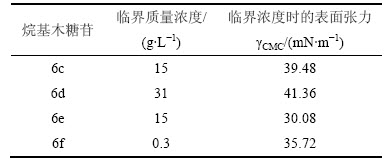
��4 ���-��-D-���ľ����6c~6f����Ũ��Ϊ0.3 g/Lʱ��Ӧ�ı�������
Table 4 Surface tension of xylopyranoside 6c~6f when concentration of 0.3 g/L


ͼ9 ľ����(6a~6k)����������ʧ�ʷ���
Fig. 9 Thermogravimetric analysis (TGA) of xylopyranoside (6a~6i)
2.8 ���-��-D-���ľ���յ�����Һ����
��Ϊ�������ʡ�(soft matter)������֬��ͨ������װ������֯�����γɳ�����Һ�������Ķ�̬�仯������Ĥ���������﹦�����ã�����������֬���������Һ�����ܵ���Ȼ��֬��Ϊϸ��Ĥ�������������Ժͼ����ij����й�[9,19,24]�����о�����ƫ������ֱ�ӿ��첻ͬ���������-��-D-���ľ����6a~6i������Һ����Ϊ[8]�������ľ���մ���Һ����ʱ������������Ӽ�����ò�������װ������֯ģʽ�γ����ǿ���õ��ǻ������ˮ�����õ������������������У��Է���װ�ɲ�״�����ơ���Ƕ��ƽ�����е�֯����
����Һ�������Ļ���������ȶ�������ν�ġ�˫����ת�䡱���������[9]�������ȴﵽ���۵�(Mp)ʱ���ᾧ�ԵĹ�̬����ת��ɰ�����Һ��̬�������ڽϸߵ��¶�(Cp)��ת��ɸ���ͬ�Ե�������Һ̬����5��ʾΪľ���յ���ת���¶ȡ��ӱ�5�ɼ������-��-D-���ľ��������һͷһβ(one head one tail)��һͷ��һ��(one head group-one chain)�ҳ��ֶ�̬�Ե�˫��������շ���[19]���ڼ���ʱ����䷢������Խ�С���¶����䣬����������Һ���࣬�ҵ���ˮ�����̼������nΪ6ʱ�����-��-D-���ľ��������¶�֮��(��t)�ϴ������Ķ��ȽϽӽ�����ľ�����У������������������ȡ����ȶ��ԣ��������ȶ��Զ�Һ����������DZ�Ҫ�ġ����������-��-D-���ľ����Ϊ��״������ֻ��3�������ǻ��������-��-D-�����������������ˮ��ͷ�����ǻ��ϵ��Ǽ���Ҳ��ľ�Ǵ���ʮ�������������1��������ǻ���������Cp���ߣ����ԣ��õ������-��-D-���ľ���վ���Һ���࣬������¶ȷ�Χ(��t=Cp-Mp)��խ��Һ������ȶ��Խ�����
��5 ľ���յ���ת���¶�
Table 5 Phase transition temperatures of xylopyranoside

3 ����
1) ���������ǰ��������и߶�����ѡ���ԡ��˷�������Ƿ����ڵ��ؽ�����Ⱦ�ıˣ����ô˷�����ľ��Ϊԭ�ϣ�ͨ��ȫ��������ѡ����1-λ�ѱ�����ת������������ǰ�����ż�����ѱ�������5����Ӧ���ϳ���9�����-��-D-���ľ����(6a~6i)����ϵͳ�زⶨ�����ܽ��ԡ��ܽ��ʡ������������黯�ԡ�����������ĭ�ȶ��ԡ����ȶ��Լ�����Һ���ԡ�
2) ���ϳɵ����-��-D-���ľ����(6a~6i)��ˮ�е��ܽ��ԱȽϺã�������6d(n=7)������6e(n=8)���ܽ����������6e(n=8)��6f (n=9)��ʹ���������½����ϵ�ֵ��������Խ�ǿ������6e(n=8)ʹ�����������͵����ֵ����������ǿ������6f (n=9)ʹ���������½���һ��ֵʱ�����Ũ����С����Ч����ߡ�����6f (n=9)�Ա��Ͳ����͵��黯������ǿ������6e(n=8) ��6f(n=9)������������ĭ�ȶ��Ծ��Ϻá�
3) ���ϳɵ����-��-D-���ľ����(6a~6i)��150 ���������ȶ��ģ���Һ������¶ȷ�Χ��խ�����й�Ӧ�á�ϸ��������ṹ����ȹ�Ч��ϵ�д��ڽ�һ���о���
�ο����ף�
[1] HENDERSON P J F, BALOWIN S A. Bundles of insights into sugar transporters[J]. Nature, 2012, 490(7420): 348-350.
[2] TAYLOR M E, DRICKAMER K. Convergent and divergent mechanisms of sugar recognition across kingdoms[J]. Current Opinion in Structural Biology, 2014, 28: 14-22.
[3] NAGAE M, YAMAGUCHI Y. Three-dimensional structural aspects of protein�Cpolysaccharide interactions[J]. International Journal of Molecular Sciences, 2014, 15(3): 3768-3783.
[4] SATO T K, LIU T, PARREIRAS L S, et al. Harnessing genetic diversity in Saccharomyces cerevisiae for fermentation of xylose in hydrolysates of alkaline hydrogen peroxide-pretreated biomass[J]. Applied and Environmental Microbiology, 2014, 80(2): 540-554.
[5] CORREIA M A S, MAZUMDER K, BR S J L A, et al. Structure and function of an arabinoxylan-specific xylanase[J]. The Journal of Biological Chemistry, 2011, 286(25): 22510-22520.
S J L A, et al. Structure and function of an arabinoxylan-specific xylanase[J]. The Journal of Biological Chemistry, 2011, 286(25): 22510-22520.
[6] ZAHID N I, CONN C E, BROOKS N J, et al. Investigation of the effect of sugar stereochemistry on biologically relevant lyotropic phases from branched-chain synthetic glycolipids by small-angle X-ray scattering[J]. Langmuir, 2013, 29(51): 15794-15804.
[7] CHAI Jinling, WU Changju, LI Ganzuo, et al. Studies on middle-phase microemulsions of green surfactant n-dodecyl polyglucoside C12G1.46[J]. Chinese Journal of Chemistry, 2003, 21(1): 25-29.
[8] ���Ʒ�, ������, ���ΰ, ��. ���-��-D-����������յĺϳ�������[J]. Ӧ�û�ѧ, 2013, 30(10): 1120-1126.
LIU Dengfeng, CHEN Langqiu, LI Hongwei, et al. Syntheses and properties of alkyl ��-D-glucopyranosides[J]. Chinese Journal of Applied Chemistry, 2013, 30(10): 1120-1126.
[9] XU W, OSEI-PREMPEH G, LEMA C, et al. Synthesis, thermal properties, and cytotoxicity evaluation of hydrocarbon and fluorocarbon alkyl ��-D-xylopyranoside surfactants[J]. Carbohydrate Research, 2012, 349: 12-23.
[10] MATSUMURA S, SAKIYAMA K, TOSHIMA K.Preparation of octyl ��-D-xylobioside and xyloside by xylanase-catalyzed direct transglycosylation reaction of xylan and octanol[J]. Biotechnology Letters, 1999, 21(1): 17-22.
[11] KADI N, BELLOY L, CHALIER P, et al. Enzymatic synthesis of aroma compound xylosides using transfer reaction by Trichoderma longibrachiatum xylanase[J]. Journal of Agricultural and Food Chemistry, 2002, 50(20): 5552-5557.
[12] LI Y K, YAO H Y, CHO Y T. Effective induction, purification and characterization of Trichoderma koningii G-39 ��-xylosidase with high transferase activity[J]. Biotechnology and Applied Biochemistry, 2000, 31(2): 119-125.
[13] SHINOYAMA H, KAMIYAMA Y, YASUI T. Enzymatic synthesis of alkyl ��-xylosides from xylobiose by application of the transxylosyl reaction of Aspergillus niger ��-xylosidase[J]. Agricultural and Biological Chemistry, 1988, 52(9): 2197-2202.
[14] SEKINE M, KIMURA T, KATAYAMA Y, et al. The direct and one-pot transformation of xylan into the biodegradable surfactants,alkyl xylosides, is aided by an ionic liquid[J]. RSC Advances, 2013, 3: 19756-19759.
[15] SATG C, BRAS J L, HNIN F, et al. DMF promoted xylosylation of terpenols[J]. Tetrahedron, 2005, 61(35): 8405-8409.
C, BRAS J L, HNIN F, et al. DMF promoted xylosylation of terpenols[J]. Tetrahedron, 2005, 61(35): 8405-8409.
[16] PETROVI Z D, ANDJELKOVI D, SPASOJEVI A. Vitamin B12 and BF3-etherate as catalysts in synthesis of some C4-C12-alky ��-D-xylopyranosides[J]. Indian Journal of Chemistry, 2006, 45B: 272-275.
[17] KONSTANTINOVI S, PETROVI Z, SPASOJEVI A, et al. Synthesis of C7-C16-alkyl glycoside: part II. synthesis of alkyl D-xylopyranosides in the presence of tin(IV) chloride as a Lewis acid catalyst[J]. Indian Journal of Chemistry, 2001, 40B(7): 614-618.
[18] OLDHAM E D, SEELAM S, LEMA C, et al. Synthesis, surface properties, and biocompatibility of 1,2,3-triazole-containing alkyl ��-D-xylopyranoside surfactants[J]. Carbohydrate Research, 2013, 379: 68-77.
[19] GOODBY J W, G RTZ V, COWLING S J, et al. Thermotropic liquid crystalline glycolipids[J]. Chemical Society Reviews, 2007, 36(12): 1971-2032.
RTZ V, COWLING S J, et al. Thermotropic liquid crystalline glycolipids[J]. Chemical Society Reviews, 2007, 36(12): 1971-2032.
[20] ��ˮ��, �չھ�, �ؕP, ��. L-����Ѫ���¹�������ø���ϳɡ����뼰������[J]. ʳƷ��ҵ�Ƽ�, 2008, 29(10): 211-215.
CAI Shuigen, TAO Guanjun, QIN Fang, et al. Enzymatic synthesis, separation and properties of L-ascorbyl laurate[J]. Science and Technology of Food Industry, 2008, 29(10): 211-215.
[21] LI Yu, XU Li, WANG Fuan, et al. Solubilities of cefepime hydrochloride in water + (ethanol, 1-propanol, or 2-propanol) from (278.15 to 308.15)K[J]. Journal of Chemical & Engineering Data, 2010, 55(9): 4098-4103.
[22] MANI K, BELTING M, ELLERVIK U, et al. Tumor attenuation by 2(6-hydroxynaphthyl)-��-D- xylopyranoside requires priming of heparan sulfate and nuclear targeting of the products[J]. Glycobiology, 2004, 14(5): 387-397.
[23] ���¶�, �ν�, ���ľ�, ��. ʮ��������յ������о�[J]. Ӧ�û���, 2010, 39(8): 1164-1166.
DENG Yuee, SONG Jie, GONG Wenjun, et al. Study on the properties of dodecyl polyglucosides[J]. Applied Chemical Industry, 2010, 39(8): 1164-1166.
[24] MARTIN P, GOD P, VILLA P, et al. D-xylose derivative liquid crystals[J]. Journal of Thermal Analysis and Calorimetry, 2001, 63(2): 339-344.
(�༭ �²ӻ�)
�ո����ڣ�2015-11-12�������ڣ�2015-01-15
������Ŀ(Foundation item)������ʡ��Ȼ��ѧ����������Ŀ(14JJ2067��10JJ6023)����̶��ѧ��ʮ����ѧ�����»���������Ŀ(2014xtuxj31)(Projects(14JJ2067, 10JJ6023) supported by the Natural Science Foundation of Hunan Province; Project(2014xtuxj31) supported by the Tenth College Students Innovation Fund of Xiangtan University)
ͨ�����ߣ���������ڣ������ǻ�ѧ���л��ϳɡ�ҩ��ϳɺ�ʳƷ���Ӽ��о���E-mail��chengood2003@263.net

