Trans. Nonferrous Met. Soc. China 23(2013) 1718-1722
Synthesis and performance of LiVPO4F/C-based cathode material for lithium ion battery
Jie-xi WANG, Zhi-xing WANG, Li SHEN, Xin-hai LI, Hua-jun GUO, Wei-jia TANG, Zhen-guo ZHU
School of Metallurgy and Environment, Central South University, Changsha 410083, China
Received 25 July 2012; accepted 17 January 2013
Abstract: LiVPO4F/C-based material was prepared by heating a precursor, which was obtained through ball milling with slurry of H2C2O4・2H2O, NH4H2PO4, NH4VO3 and LiF. In this process, H2C2O4・2H2O was used as a reducing agent as well as carbon source. The properties of the precursor and the prepared sample were characterized by TG-DTA, XRD, SEM, TEM and C-S analysis. XRD analysis shows that the precursor is amorphous and the prepared LiVPO4F sample coexists with Li3V2(PO4)3, as well as a small amount of V2O3. Homogeneous powders with an average particle size of 2 μm are observed by TEM. HRTEM image shows that the crystal particles are coated by amorphous carbon uniformly, with a coating layer of about 2 nm. The synthesized material shows the specific discharge capacities of 151.3 and 102.5 mA・h/g at 0.1C and 10C respectively, in the voltage range of 3.0-4.4 V, and it keeps 90.4% of initial discharge capacity at 10C after 50 cycles. V3+/V4+ redox peaks corresponding to both LiVPO4F and Li3V2(PO4)3 are seen clearly in the CV curve.
Key words: lithium ion battery; LiVPO4F; ball milling; cathode material; carbon coating
1 Introduction
Framework materials based on the phosphate polyanion, such as LiFePO4, LiMnPO4 and Li3V2(PO4)3, have been firmly believed to be potential electro-active materials for lithium-ion battery applications [1-7]. Fluorophosphate, represented by the general formula RMPO4F, where R represents lithium or sodium and M represents a 3d transition metal, was reported as electro- active material by BARKER et al [8,9]. LiVPO4F, namely lithium vanadium fluorophosphate, owing to its good electrochemical performance, high voltage platform, high energy density and excellent thermal stability, is considered to a novel 4 V cathode material to substitute LiCoO2 commonly used in the first-generation lithium-ion batteries [10,11]. It is isostructural with the naturally occurring minerals tavorite LiFePO4・OH, or amblygonite LiAlPO4F, crystallizing with a triclinic structure (space group 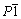 ) [12-14]. The reversible Li+ extraction/insertion reaction for Li1-xVPO4F based on the V3+/4+ redox couple operates at about 4.2 V versus Li, which is 0.2 or 0.7 V higher than that exhibited by LiCoO2 or LiFePO4, respectively [11,15]. Usually, LiVPO4F is synthesized by conventional two-step carbon thermal reaction (CTR), which generally operates at high temperatures for a long time. BARKER et al [8] first introduced the two-step CTR method for preparing LiVPO4F/C successfully, and did much to improve its electrochemical properties by two-step CTR method [10,16,17]. Unfortunately, though the utilization of lithium ion is increased, the poor rate performance still hinders its further development as the next commercial high-potential cathode material. One-step CTR method is also employed to prepare LiVPO4F/C. ZHONG et al [18] employed humic acid as reduction agent as well as carbon source, and prepared porous LiVPO4F/C particles by one-step method successfully. A sol-gel method was also employed to synthesize LiVPO4F. LI et al [19] reported a novel sol-gel method to prepare LiVPO4F/C, using H2O2 as coordinating agent to prepare V2O5・nH2O precursor, which has an important significance. ZHONG et al [15] and QIAO et al [20] employed citric acid to prepare Li-V-PO4-F gel and then sintered it at high temperatures to obtain crystal LiVPO4F [15,20]. But it may not be an effective way for large scale production.
) [12-14]. The reversible Li+ extraction/insertion reaction for Li1-xVPO4F based on the V3+/4+ redox couple operates at about 4.2 V versus Li, which is 0.2 or 0.7 V higher than that exhibited by LiCoO2 or LiFePO4, respectively [11,15]. Usually, LiVPO4F is synthesized by conventional two-step carbon thermal reaction (CTR), which generally operates at high temperatures for a long time. BARKER et al [8] first introduced the two-step CTR method for preparing LiVPO4F/C successfully, and did much to improve its electrochemical properties by two-step CTR method [10,16,17]. Unfortunately, though the utilization of lithium ion is increased, the poor rate performance still hinders its further development as the next commercial high-potential cathode material. One-step CTR method is also employed to prepare LiVPO4F/C. ZHONG et al [18] employed humic acid as reduction agent as well as carbon source, and prepared porous LiVPO4F/C particles by one-step method successfully. A sol-gel method was also employed to synthesize LiVPO4F. LI et al [19] reported a novel sol-gel method to prepare LiVPO4F/C, using H2O2 as coordinating agent to prepare V2O5・nH2O precursor, which has an important significance. ZHONG et al [15] and QIAO et al [20] employed citric acid to prepare Li-V-PO4-F gel and then sintered it at high temperatures to obtain crystal LiVPO4F [15,20]. But it may not be an effective way for large scale production.
In this work, a simple method is presented to synthesize carbon coated LiVPO4F, namely chemical reduction assisted with mechanical activation followed by short-time calcinations. The V(+5) is reduced to V(+3) by oxalic acid at room temperature and it is not necessary to prepare VPO4 intermediate at high temperatures compared with traditional CTR method. The short-time calcination can save a large amount of energy. The carbon is decomposed from gas-phase reaction and can be deposited on the surface of the crystal particle uniformly. The electrochemical performance of prepared samples is evaluated.
2 Experimental
The LiVPO4F sample was prepared as following steps. Firstly, stoichiometric NH4VO3 (AR, 99%), NH4H2PO4 (AR, 98.5%), LiF (AR, 99.5%), 50% excess oxalic acid dihydrate (H2C2O4・2H2O, AR, 98.5%) were added to a sealing can, and ball milled in the alcohol media at a revolving speed of 250 r/min for 4 h. The ball mill type was ND6-2L with rated power of 0.75 kW. The oxalic acid dihydrate here was introduced as reducing agent as well as carbon source. Secondly, the obtained slurry was dried in a drying oven at 80 °C for 6 h. Finally, the dried powders were sintered at 350 °C for 5 h, and then at 750 °C for 90 min under argon atmosphere.
The TG-DTA analysis was tested by SDTQ600 with a step time of 10 °C/min under argon atmosphere. The powder X-ray diffraction (XRD, Rint-2000, Rigaku) using Cu Kα radiation was employed to identify the crystalline phase of the synthesized material. The mass fractions of the crystal phases were evaluated by multiphase refinement with Maud Program. The morphology of the LiVPO4F powders was observed on a scanning electron microscope (SEM, JEOL, JSM- 5612LV) with an accelerating voltage of 20 kV, and on a transmission electron microscope (TEM, Tecnai G12). Carbon content was tested by a C-S analyzer (Eltar, Germany).
The electrochemical characterizations were conducted with CR2025 coin-type cell. For positive electrode fabrication, the prepared powders were mixed with 10% acetylene black and 10% polyvinylidene fluoride in N-methyl pyrrolidinone until slurry was obtained. And then, the blended slurry was pasted onto an aluminum current collector, followed by drying at 120 °C for 12 h in the air. The test cell consisted of the positive electrode and lithium foil negative electrode separated by a porous polypropylene film, using 1 mol/L LiPF6 in EC, EMC and DMC (1:1:1 in volume) as the electrolyte. The assembly of the cells was carried out in a dry Ar-filled glove box. Electrochemical tests were carried out using an automatic galvanostatic charge- discharge unit, NEWARE battery circler, between 3.0 V and 4.4 V versus Li/Li+ at room temperature. A constant current-constant voltage condition was employed for charge process and a galvanostatic condition was conducted for discharge process. The cyclic voltammetry measurement at a scan rate of 0.05 mV/s between 3.0 V and 4.4 V was carried out with a CHI660D electrochemical analyzer.
3 Results and discussion
In the process of ball milling for raw materials NH4VO3, NH4H2PO4, LiF and H2C2O4・2H2O at room temperature, the pentavalent vanadium is reduced to trivalent vanadium by H2C2O4・2H2O, and the color of the material changes from creamy white to green. Figure 1(a) shows the XRD pattern of LiVPO4F precursor prepared by ball milling at room temperature. It can be seen that there is no obvious evidence of diffraction peaks in Fig. 1(a), which indicates that the precursor is nearly amorphous. The TG-DTA curves of the precursor are shown in Fig. 1(b). It is clear that three endothermic peaks and mass loss stages appear at 75-135 °C, 250-390 °C and 450-700 °C, which represent the loss of absorbed water, decomposition of oxalic acid as well as ammonium dihydrogen phosphate, formation accompanied crystallization of LiVPO4F, respectively. Two distinct changes are observed at 450 and 630 °C. The change at 450 °C may correspond to further decomposition of intermediate product H3PO4 and H4P2O7 to (HPO3)x and H2O(g), and the mass loss at 630 °C may result from formation of crystal LiVPO4F, in which the volatilization of H2O(g) may induce HF sublimation. According to the above analysis, two-step calcination schedule should be introduced to ensure sufficient decomposition and contact among the reactants, and finally materials with good electrochemical performance can be obtained.
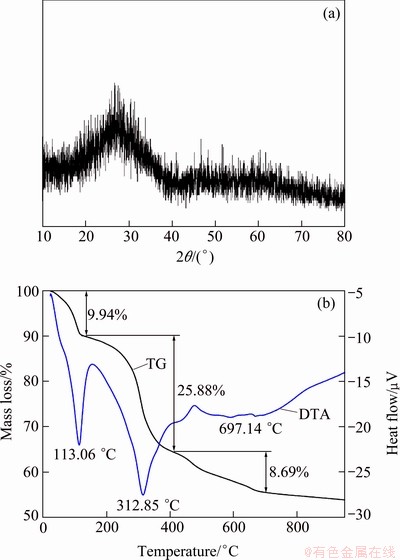
Fig. 1 XRD pattern (a) and TG-DTA curves (b) of LiVPO4F precursor
Figure 2 shows the XRD pattern of the prepared material after heat treatment under argon atmosphere. The XRD pattern shows a series of diffraction peaks, indicating that a transformation of the precursor from amorphous to a crystalline phase occurs during the calcination process. A multiphase refinement method was employed to analyze the XRD based on the models of LiFePO4(OH,F) (ICSD #20808), Li3V2(PO4)3 (ICSD #98362) and V2O3 (ICSD #1473). The result shows that the mass fractions of LiVPO4F, Li3V2(PO4)3 and V2O3 are 79.2%, 17.6%, 3.2%, respectively. Generally, the fitting inequality level factors Rw value should be less than 15%. Herein, the value of Rw is 8.86%, indicating the refined results are credible. The existence of Li3V2(PO4)3 in the prepared material may be on account of reactions between fluorochemicals and H2O(g) at high temperatures, causing formation of HF. The volatilization of HF results in decrease of fluorine and vanadium contents in solid phase, causing the formation of Li3V2(PO4)3.
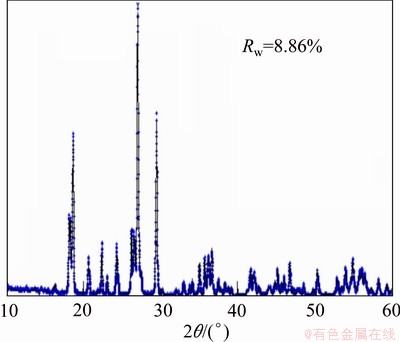
Fig. 2 XRD pattern of prepared material after heat treatment
Figure 3(a) shows the SEM image of LiVPO4F sample. In the micrograph, particles with the average size of about 2 μm show good crystallinity and uniformity and the grains are even compared with those synthesized by sol-gel method [19]. The morphology and surface state of the prepared LiVPO4F particles were observed by TEM and HRTEM, as shown in Figs. 3(b) and (c). From the result, it can be seen that a carbon layer of about 2 nm adheres on the surface of the prepared particles. The fast Fourier transform (FFT) (inset in Fig. 3(c)) shows the lattice planes according to (110) and (010) planes of LiVPO4F crystal. C-S analysis shows that the carbon content is about 3.7%. The carbon derives from the decomposition of excessive oxalic acid dihydrate (H2C2O4・2H2O), and is helpful to electronic conductivity during electrode process. It is worth taking a moment to flag the fact that H2C2O4 is decomposed to C and CO2 at low temperature indirectly, which can be described as the following chemical equation:
H2C2O4=CO(g)+CO2(g)+H2O(g) (1)
2CO(g)=C(s)+CO2(g) (2)
When the temperature is lower than 400 °C, CO gas is unstable and decomposes to C(s) and CO2 gas. But the stability of CO increases with elevating the temperature. So, it is necessary to keep a period of time at lower temperatures to ensure production of carbon.

Fig. 3 SEM (a), TEM (b) and HRTEM (c) images of prepared sample after heat treatment
Figure 4(a) shows the discharge profiles of the LiVPO4F/C cathode at different rates. In the discharge curves, there is a long plateau at about 4.2 V, which corresponds to Li+ insertion in Li1-xVPO4F (0≤x≤1). A short plateau at about 4.0 V following the long one indexes with the reduction of V4+ to V3+ in Li3-xV2(PO4)3 (1≤x≤2). Two short continuous plateaus at 3.7-3.5 V are in accordance with Li+ further insertion into Li3-xV2(PO4)3 (0≤x≤1). From Fig. 4(a), it can be seen that the initial discharge capacity of LiVPO4F sample is about 151.3 mAh/g at 0.1C (1C equals to 150 mA/g). Judged from the plateaus of the discharge curve, the capacity is about 120 mA・h/g delivered by LiVPO4F and 30 mA・h/g displayed by Li3V2(PO4)3, which is consistent with the XRD analysis of prepared sample, as shown in Fig. 2. And the utilization of the active material decreases by the increase of rate, performing 150.9, 147.6, 122.8, and 102.5 mA・h/g, at 0.5C, 2C, 5C and 10 C, respectively. The above results appear excellent compared with those synthesized through traditional two-step CTR method [8,12] and sol-gel method [15]. The prepared material possesses a relatively good cycle performance at a high discharge rate and the capacity retention keeps 90.4% after 50 cycles at 10C, as shown in Fig. 4(b).
Fig. 4 Initial discharge curves (a) and cycle performance (b) at various rates
The relatively high capacity and good cycle performance of the material may be attributed to structural stability of LiVPO4F [14]. Due to the strong electro-negativity of fluorine, the addition of fluorine atoms makes the PO4 tetrahedron and VO4F2 octahedron more stable, thus offering more pourable passageways for Li+ insertion/extraction. It is also attributed to the micro-size particles and carbon coating. The small particles have a large contact area between the electrode material and electrolyte solution in an electrochemical cell. As is well known, the coated carbon is a favorable conductive agent, and it can obviously improve the electronic conductivity in the electrochemical process. In addition, Li3V2(PO4)3 impurity may affect the electro- chemical performance of the prepared materials [21,22].
For further investigating the effects of Li3V2(PO4)3 on the electrochemical performance of the prepared electrode, a cyclic voltammogram (CV) test was conducted at the scan rate of 0.05 mV/s between 3.0 and 4.4 V. From the CV curves in Fig. 5, V3+/V4+ redox peaks for both Li3-xV2(PO4)3 and Li1-xVPO4F are observed clearly in their curves, which are consistent with the initial charge-discharge test. This indicates that two independent electro-active materials, LiVPO4F and Li3V2(PO4)3, coexist in the prepared sample. The peaks for Li1-xVPO4F are sharper than those for Li3-xV2(PO4)3, indicating that LiVPO4F is the main capacity contributor.
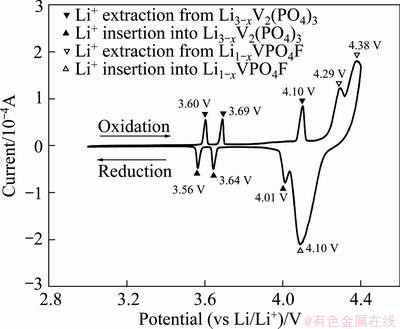
Fig. 5 CV curve of prepared sample at scan rate of 0.05 mV/s
4 Conclusions
1) Carbon coated LiVPO4F, coexisting with Li3V2(PO4)3 and V2O3, is prepared by chemical reduction assisted with mechanical activation followed by short-time calcination. The oxalic acid is used as reducing agent as well as carbon source, and the pentavalent vanadium is reduced to trivalent vanadium by oxalic acid.
2) The synthesized crystal particles are adhered by an amorphous carbon layer. The high electronic conductive carbon film is deposited from CO at low temperatures, which is decomposed from the excess oxalic acid.
3) Both LiVPO4F and Li3V2(PO4)3 act as electro-active materials and contribute to the capacity.
4) The prepared sample possesses good electrochemical performance, which is attributed to the stable structure of LiVPO4F, micro-size particles and coated carbon layer.
References
[1] FISHER C A J, HART PRIETO V M, ISLAM M S. Lithium battery materials LiMPO4 (M=Mn, Fe, Co, and Ni): Insights into defect association, transport mechanisms, and doping behavior [J]. Chemistry of Materials, 2008, 20(18): 5907-5915.
[2] CHEN Han, YU Wen-zhi, HAN Shao-chang, XU Zhong-yu. Preparation and electrochemical properties of LiFePO4/C composite with network structure for lithium ion batteries [J]. Transactions of Nonferrous Metals Society of China, 2007, 17(5): 951-956.
[3] WU Ling, WANG Zhi-xing, LI Xin-hai, LI Ling-jun, GUO Hua-jun, ZHENG Jun-chao, WANG Xiao-juan. Electrochemical performance of Ti4+-doped LiFePO4 synthesized by co-precipitation and post-sintering method [J]. Transactions of Nonferrous Metals Society of China, 2010, 20(5): 814-818.
[4] ZHANG Bao, LIU Jie-qun, ZHANG Qian, LI Yan-hong. Electrochemical performance of Al-substituted Li3V2(PO4)3 cathode materials synthesized by sol-gel method [J]. Transactions of Nonferrous Metals Society of China, 2010, 20 (4): 619-623.
[5] ZHANG Qian, LI Yan-hong, ZHONG Sheng-kui, XIAO Xin-he, YAN Bo. Synthesis and electrochemical performance of Li3V2(PO4)3 by optimized sol-gel synthesis routine [J]. Transactions of Nonferrous Metals Society of China, 2010, 20(8): 1545-1549.
[6] ZHONG Sheng-kui, WANG You, LIU Jie-qun, WANG Jian. Synthesis of LiMnPO4/C composite material for lithium ion batteries by sol-gel method [J]. Transactions of Nonferrous Metals Society of China, 2012, 22(10): 2535-2540.
[7] HE Li-hua, ZHAO Zhong-wei, LIU Xu-heng, CHEN Ai-liang, SI Xiu-fen. Thermodynamics analysis of LiFePO4 pecipitation from Li-Fe(II)-P-H2O system at 298 K [J]. Transactions of Nonferrous Metals Society of China, 2012, 22(7): 1766-1770.
[8] BARKER J, SAIDI M Y, SWOYER J L. Electrochemical insertion properties of the novel lithium vanadium fluorophosphate, LiVPO4F [J]. Journal of the Electrochemical Society, 2003, 150(10): A1394-A1398.
[9] BARKER J, SAIDI M Y, SWOYER J L. A sodium-ion cell based on the fluorophosphate compound NaVPO4F [J]. Electrochemical and Solid-State Letters, 2003, 6(1): A1-A4.
[10] GOVER R K B, BURNS P, BRYAN A, SAIDI M Y, SWOYER J L, BARKER J. LiVPO4F: A new active material for safe lithium-ion batteries [J]. Solid State Ionics, 2006, 177(26-32): 2635-2638.
[11] REDDY M V, SUBBA R G V, CHOWDARI B V R. Long-term cycling studies on 4V-cathode, lithium vanadium fluorophosphate [J]. Journal of Power Sources, 2010, 195(17): 5768-5774.
[12] BARKER J, GOVER R K B, BURNS P, BRYAN A, SAIDI M Y, SWOYER J L. Structural and electrochemical properties of lithium vanadium fluorophosphate, LiVPO4F [J]. Journal of Power Sources, 2005, 146(1-2): 516-520.
[13] ELLIS B L, RAMESH T N, DAVIS L J M, GOWARD G R, NAZAR L F. Structure and electrochemistry of two-electron redox couples in lithium metal fluorophosphates based on the tavorite structure [J]. Chemistry of Materials, 2011, 23(23): 5138-5148.
[14] ATEBA MBA J M, MASQUELIER C, SUARD E, CROGUENNEC L. Synthesis and crystallographic study of homeotypic LiVPO4F and LiVPO4O [J]. Chemistry of Materials, 2012, 24(6): 1223-1234.
[15] ZHONG Sheng-kui, CHEN Wei, LI Yan-hong, ZOU Zheng-guang, LIU Chang-jiu. Synthesis of LiVPO4F with high electrochemical performance by sol-gel route [J]. Transactions of Nonferrous Metals Society of China, 2010, 20(s1): s275-s278.
[16] BARKER J, SAIDI M Y, SWOYER J L. A comparative investigation of the Li insertion properties of the novel fluorophosphate phases, NaVPO4F and LiVPO4F [J]. Journal of the Electrochemical Society, 2004, 151(10): A1670-A1677.
[17] BARKER J, GOVER R K B, BURNS P, BRYAN A J. A lithium-ion cell based on Li4/3Ti5/3O4 and LiVPO4F [J]. Electrochemical and Solid-State Letters, 2007, 10(5): A130-A133.
[18] ZHONG Sheng-kui, WANG Jian, LI Yan-wei, LIU Le-tong, LIU Jie-qun, YANG Jiang-wei. One-step synthesis of LiVPO4F/C cathode material with high performance [J]. Chemistry Letter, 2009, 38(4): 374-375.
[19] LI Yu-zhan, ZHOU Zhen, GAO X P, YAN Jie. A novel sol-gel method to synthesize nanocrystalline LiVPO4F and its electrochemical Li intercalation performances [J]. Journal of Power Sources, 2006, 160(1): 633-637.
[20] QIAO Xiao-chang, YANG Jian-dong, WANG Yao-bin, CHEN Quan-qi, ZHANG Ting-ting, LIU Li, WANG Xian-you. Electrochemical performance of LiVPO4F/C composite cathode prepared through amorphous vanadium phosphorus oxide intermediate [J]. Journal of Solid State Electrochemistry, 2012, 16(3): 1211-1217.
[21] ZHONG Sheng-kui, WU Ling, LIU Jie-qun. Sol-gel synthesis and electrochemical properties of 9LiFePO4・Li3V2(PO4)3/C composite cathode material for lithium ion batteries [J]. Electrochimica Acta, 2012, 74: 8-15.
[22] WANG Jie-xi, WANG Zhi-xing, LI Xin-hai, GUO Hua-jun, WU Xian-wen, ZHANG Xiao-ping, XIAO Wei. xLi3V2(PO4)3・LiVPO4F/C composite cathode materials for lithium ion batteries [J]. Electrochimica Acta, 2013, 87: 224-229.
锂离子电池用LiVPO4F/C 基正极材料的制备和性能
王接喜,王志兴,沈 力,李新海,郭华军,唐维佳,朱振国
中南大学 冶金科学与工程学院,长沙 410083
摘 要:将H2C2O4・2H2O, NH4H2PO4, NH4VO3 和 LiF通过球磨反应、烧结,合成了 LiVPO4F/C 基正极材料。在这个过程中,草酸起还原剂和碳源的作用,利用热重、 X射线衍射、 扫描电镜、透射电镜和碳-硫分析等手段对合成的前驱体和材料进行检测和分析。XRD分析表明,球磨反应后所得到的前驱体为无定形态,而烧结后的材料中除了LiVPO4F的衍射峰外,还存在Li3V2(PO4)3和V2O3衍射峰。材料颗粒均匀,尺寸约2 μm。透射电镜分析表明,合成的材料颗粒表面包裹着一层约2 nm厚的无定形碳。在截止电压3.0~4.4 V时,合成的材料在0.1C和10C倍率下的放电比容量分别为151.3和102.5 mA・h/g。在10C倍率下循环50次后容量保持率为90.4%。在LiVPO4F 和Li3V2(PO4)3 的循环伏安曲线中可以明显看到V3+/V4+的氧化还原峰。
关键词:锂离子电池;LiVPO4F;球磨;正极材料;碳包覆
(Edited by Xiang-qun LI)
Foundation item: Project (2011FJ1005) supported by Science and Technology Major Projects of Hunan Province, China; Project supported by Hunan Provincial Innovation Foundation for Postgraduate, China
Corresponding author: Zhi-xing WANG; Tel/Fax: +86-731-88836633; E-mail: zxwang.csu@hotmail.com
DOI: 10.1016/S1003-6326(13)62653-9

