
Crystal structure and negative thermal expansion properties of solid solution Er2W3-xMoxO12
PENG Jie(彭 杰)1, 2, LIU Xin-zhi(刘新智)3, GUO Fu-li(郭富丽)4, HAN Song-bai(韩松柏)3, LIU Yun-tao(刘蕴韬)3, CHEN Dong-feng(陈东风)3, ZHAO Xin-hua(赵新华)4, HU Zhong-bo(胡中波)2
1. Experimental Physics Center, Institute of High Energy Physics, Chinese Academy of Sciences,
Beijing 100049, China;
2. College of Chemistry and Chemical Engineering, Graduate University of the Chinese Academy of Sciences,
Beijing 100049, China;
3. China Institute of Atomic Energy, Beijing 102413, China;
4.College of Chemistry, Beijing Normal University, Beijing 100875, China
Received 10 August 2009; accepted 15 September 2009
Abstract: A series of solid solutions Er2W3-xMoxO12 (0.5≤x≤2.5) were successfully synthesized by the solid state method. Their crystal structures and negative thermal expansion properties were studied by high temperature X-ray powder diffraction and the Rietveld method. All samples with rare earth tungstates and molybdates crystallize in the same orthorhombic structure with space group Pnca, and show the negative thermal expansion phenomena related to transverse vibration of bridging oxygen atoms in the structure. Thermal expansion coefficients (TECs) of Er2W3-xMoxO12 were determined as -16.2×10-6 K-1 for x=0.5 and -16.5×10-6 K-1 for x=2.5 while -20.2×10-6 K-1 and -18.4×10-6 K-1 for unsubstituted Er2W3O12 and Er2Mo3O12 in the identical temperature range of 200-800 ℃. High temperature XRD data and bond length analysis suggest that the difference between W-O and Mo-O is responsible for the change of TECs after the element substitution in the series of solid solutions.
Key words: negative thermal expansion; X-ray diffraction; rare earth; molybdate; tungstate
1 Introduction
Negative thermal expansion (NTE) was reported in a large family of tungstates and molybdates with the general formula A2M3O12, where A could be Sc, Y and lanthanide rare earth elements Ho, Er, Tm, Yb and Lu[1-10]. This type of compounds crystallizes in space group Pnca. Their structure consists of a corner-sharing network of AO6 octahedra and W(Mo)O4 tetrahedra. The AO6 octahedra shares corners with six W(Mo)O4 tetrahedra while W(Mo)O4 tetrahedra shares corners with four AO6 octahedra[1, 3-4, 8-9], that is, each oxygen atom is coordinated with one +3 cation and one +6 W/Mo cation. The mechanism of their NTE has been explained in terms of transverse vibration of the bridging oxygen atom between the two rigid polyhedra[1]. It has been suggested by FORSTER and SLEIGHT[3] that the magnitude of NTE in this family is related to cation size. Larger cations expand the octahedron, reducing oxygen-oxygen repulsion within the polyhedron. This facilitates polyhedra shape changes, which is necessary for the rocking motions required by NTE[10].
All of these A2M3O12 phases are hygroscopic at room temperature[5-6, 8-10]. Since erbium has the largest ionic radius (0.89 ?) among lanthanide rare earth elements[11-12], Er2W3-xMoxO12 (0≤x≤3.0) series were chosen due to the fact that there are less hygroscopic compounds within the Sc2W3O12 type structure[9, 13]. The pure Er2W3O12 and Er2Mo3O12 have different TECs of -20.2×10-6K-1 and -18.4×10-6K-1, respectively[8-9], although W3+ (0.42 ?) and Mo3+ (0.41 ?) have the same crystal structures and almost the same ionic radii[11]. Also, little work had previously been done on substitution of position M in the A2M3O12-type compounds except the Al2Mo3-xWxO12 series. But it was found that substitution of W for Mo could affect structural and thermal expansion properties of the unsubstituted phases[14-15]. So, in this work we explore changes in the structure and thermal expansion properties of solid solution Er2W3-xMoxO12 (0≤x≤3.0).
2 Experimental
Er2W3-xMoxO12 (0.5≤x≤2.5) solid solutions were synthesized through the conventional solid state route from corresponding oxides Er2O3 (purity 99.99%), MoO3 (purity≥99.5%) and WO3 (purity≥99.0%). The reactants were preheated at 500℃ for 4 h before thorough mixing in stoichiometric proportion, then calcined at 700 ℃ for 12 h, followed by sintering at 1 000 ℃ for 24 h then at 1 100 ℃ for 24 h, with regrinding between two processes. All samples were slowly cooled to room temperature in furnace.
Room temperature XRD data were collected on a MSAL-XD2 powder diffractometer with CuKα radiation in the 2θ range of 10?-70? by step scanning. High- temperature powder XRD data were collected on a Panalytical X’pert Pro MPD with an Anton Parr high-temperature attachment also using Cu Kα (λ= 1.540 56 ?, 40 kV, 40 mA). A platinum heater was used as sample stage. The temperature was controlled at an accuracy of 1℃ by an Eurotherm temperature programmer. The heating speed was 30 ℃/min and data were collected after temperature was kept constant for 5 min. Rietveld refinements of the diffraction data were performed using the program suite Fullprof[16].
3 Results and discussion
3.1 Room temperature XRD data and structure
Phase identification of the samples with different Mo contents was carried out through X-ray diffraction at room temperature. Fig.1 shows the XRD data of Er2W3-xMoxO12 (x=0.5, 1.0, 1.5, 2.0, 2.5) from 10? to 35? to show changes in diffraction patterns. Rough Rietveld analyses of the XRD data indicate that all samples were single phase but were hygroscopic and crystallized in orthorhombic form but for x=1.5, which was judged from peak splitting and confirmed as a mixture of Er2W3O12 and Er2Mo3O12 by Rietveld refinement. From Fig.1 it is found the crystal structure changes much with higher level of Mo substitution, including peak shifts and intensity changes. All these changes result from variation of the cell parameters and shifts in atom displacements within the unit cell, even the presence of water molecules. Also, it was verified that these Er series compounds are more hygroscopic than other heavy rare earth elements lanthanides. Rietveld refinements indicate significant changes appearing in these compounds. For the reason that erbium has almost the largest ionic radius, molybdate and tungstate have the most open structure. Therefore, there is high special space for water molecule to enter the crystal lattice.

Fig.1 X-ray diffraction patterns of solid solution Er2W3-xMoxO12 at room temperature
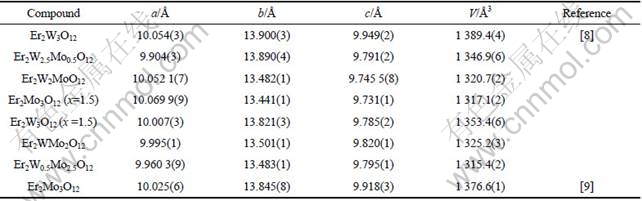
The rough refined lattice parameters of Er2W3-xMoxO12 (0.5≤x≤2.5) solid solutions at room temperature are given in Table 1. For comparison, cell parameters of unhydrated Er2W3O12 and Er2Mo3O12 are included[8-9]. As mentioned above, lattice parameters a, b, c and unit cell volume V change with no obvious trend though the ionic radius of Mo3+ is smaller than that of W3+. The dependence of lattice parameters on molybdenum content does not follow the Vegard’s law [17-18], confirming the presence of water. By comparing with the unhydrated phase[8-9], cell parameters a, b, c and unit volume V of Er2W3O12 phase in the x=1.5 mixture decrease, which is probably due to the presence of water molecules[19]. For Er2Mo3O12, other than the cell parameter a which increases from 10.025(6) to 10.069 9(9) ?, both b and c decrease, and the unit cell volume decreases from 1 376.6(1) ?3 to 1 317.1(2) ?3. It is then calculated that after getting rid of water molecules, the cell volumes increase by 2.7% and 4.5%, respectively, for Er2W3O12 and Er2Mo3O12. SUMITHRA and UMARJI[5] found that the cell volume of the hydrated pattern is 7% smaller than the unhydrated cell volume for Y2W3O12. It could be concluded that the Er2Mo3O12 is easier to absorb water molecules than Er2W3O12, and Er2W3O12 is less hygroscopic than Y2W3O12. By comparing the cell parameters of Er2W1.0Mo2.0O12 with those of Er2W0.5Mo2.5O12, it was found that a, b, c and V all decrease when W is substituted by Mo atom. This observation is understood as a consequence of Mo occupying W position in the crystal lattice. Because of the difference in bond length, the lattice becomes small with Mo substitution.
Table 1 Lattice parameters of Er2W3-xMoxO12 (x=0.5, 1.0, 1.5, 2.0, 2.5) at room temperature
3.2 High temperature XRD and negative thermal expansion
To study the effects of M site cation substitution on thermal expansion properties, high temperature X-ray powder diffraction data were collected for Er2W3-xMoxO12 (x=0.5, 2.5) at 200, 400, 600 and 800 ℃. As an example, the observed and calculated XRD patterns of Er2W2.5Mo0.5O12 at 800 ℃ and their difference are illustrated in Fig.2. It can be judged that the refinement result is very good. Also none of the high temperature XRD patterns of Er2W2.5Mo0.5O12 shows any substantial change in the experimental temperature range of 200-800 ℃ (Fig.3), which indicates that the compound structures kept unchanged and no phase transition was observed in the selected range. By contrasting with room temperature diffraction profile (Fig.1), it can be found much difference between peak intensity and profile. However, almost all the peaks shift towards high angle range with increasing temperature. For example, peaks (121), (131), (123) according to the Bragg equation indicate the existence of negative thermal expansion behavior. Cell parameters and unit cell volume values at different temperatures are listed in Table 2. Due to the complication of water molecules staying in samples at low temperature, only the data from 200 to 800 ℃ are considered. It is seen from Table 2 that cell parameters a, b, c and V of Er2W3-xMoxO12 (x=0.5, 2.5) decrease with increasing temperature in the whole experiment range, showing strong NTE behavior.

Fig.2 XRD patterns of Er2W2.5Mo0.5O12 at 800 ℃ (Rp=5.51%, Rwp=7.13% and Rexp=1.81%: (a) Calculated data; (b) Raw data; (c) Difference

Fig.3 X-ray diffraction patterns of Er2W2.5Mo0.5O12 at different temperatures
Table 2 Crystallographic data of Er2W3-xMoxO12 (x=0.5, 2.5) solid solutions at 200, 400, 600 and 800 ℃

The thermal expansion coefficients can be expressed by the following equations[20]:
 ,
, 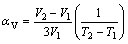 (1)
(1)
where αl and αV denote axial and volume TECs; li denote the cell parameter a or b or c; V is the unit cell volume. Because no linear relationship is observed for most of the cell parameters, the average axial and volume TECs are calculated and listed in Table 3. TECs of unsubstituted phase Er2W3O12 and Er2Mo3O12 in the same temperature range of 200-800 ℃ are also tabulated for comparison. From Table 3, it can be seen that when x=0.5, that is, Mo is used to replace W, the TECs of αa, αb, αc change from -10.14×10-6 to -8.67×10-6 K-1, -3.35×10-6 to -1.65×10-6 K-1, -6.70×10-6 to -5.90×10-6 K-1, respectively. Therefore, substitution makes the thermal expansion of all axes less negative, leading to αV from -6.74×10-6 to -5.39×10-6 K-1. When x=2.5, substitution of W for Mo has almost same effects on the crystal axes, which makes the TEC of αa change from -7.01×10-6 to -8.87×10-6 K-1, more negative than before, while TECs along axes b and c show less negative, from -4.33×10-6 to -1.32×10-6 K-1, -7.08×10-6 to -6.40×10-6 K-1, resulting in the phenomenon that αV changes from -6.14×10-6 to -5.51×10-6 K-1. By comparing with the unsubstituted phases, substitution of Mo for W makes αV decrease by 20.0%; substitution of W for Mo makes αV decrease by 10.3%. It is concluded that different substitution elements have different effects on the NTE. With the same substitution content, Mo plays more important role than W. The results imply that substitution is a good method to adjust TEC of material. In other words, it is possible to obtain materials with desired TEC by adjusting substitution elements and content.
Table 3 Axial and volume thermal expansion coefficients of solid solutions from 200 to 800 ℃

Thermal expansion is usually considered an effect of anharmonic interatomic potential. Increasing temperature causes thermal expansion of chemical bonds, which in turn leads to expansion of crystal lattice. EVANS et al[1] proposed that negative thermal expansion of the Y2(WO4)3 structure results from transverse vibrations of two coordinate bridging oxygen atoms, which makes M-O bond length unchanged but average angle of M-O-W decrease while the two cations are closer, leading to the presence of NTE. It is seen from Table 4 that average distances between Er and W(Mo) decrease from 3.891 to 3.883 ? and 3.882 to 3.875 ?, respectively, for Er2W2.5Mo0.5O12 and Er2W0.5Mo2.5O12. In this study, the change of NTE can be attributed to variation of bond strength. For example, substitution of Mo and W makes the W(Mo)-O weaker than unsubstituted W-O and Mo-O, and the weaker bonds, in turn, decrease rigidity of WO4. The rigidity of structure could affect the NTE property[4, 10], and the more the rigid polyhedra is, the larger the NTE of compound tends to show. Therefore, in this work the substitution of Mo and W impairs the NTE property of Er2W3O12 and Er2Mo3O12.
Table 4 Non-bond distance and average distance for Er2W3-xMoxO12 (x=0.5, 2.5) at 200-800 ℃

4 Conclusions
1) Solid solutions Er2W3-xMoxO12 (0.5≤x≤2.5) were successfully prepared and characterized by Rietveld analysis of X-ray powder diffraction data. The lattice parameters do not show regular trend with increasing Mo content. When x=1.5, a mixture of two pure phases appears. Water molecule absorbed in the structure can affect cell parameters of Er2W3-xMoxO12.
2) Negative thermal expansion properties of Er2W3-xMoxO12 (x=0.5, 2.5) were studied by high-temperature X-ray diffraction. Substitution of Mo and W both weakens the NTE property of Er2W3O12 and Er2Mo3O12. It is proposed that change of W(Mo)-O bond strength affects the NTE properties through its effect on WO4 rigidity.
References
[1] EVANS J S O, MARY T A, SLEIGHT A W. Negative thermal expansion in Sc2(WO4)3 [J]. J Solid State Chem, 1998, 137: 148-160.
[2] EVANS J S O, MARY T A. Structural phase transitions and negative thermal expansion in Sc2(MoO4)3 [J]. Int J Inorg Mater, 2000, 2: 143-151.
[3] FORSTER P M, SLEIGHT A W. Negative thermal expansion in Y2W3O12 [J]. Int J Inorg Mater, 1999, 1: 123-127.
[4] WOODCOCK D A, LIGHTFOOT P, RITTER C. Negative thermal expansion in Y2(WO4)3 [J]. J Solid State Chem, 2000, 149: 92-98.
[5] SUMITHRA S, UMARJI A M. Hygroscopicity and bulk thermal expansion in Y2W3O12 [J]. Mater Res Bull, 2005, 40: 167-176.
[6] MARINKOVIC B A, JARDIM P M, AVILLEZ R R D, RIZZO F. Negative thermal expansion in Y2Mo3O12 [J]. Solid State Sci, 2005, 7: 1377-1383.
[7] XIAO X L, CHENG Y Z, PENG J, WU M M, CHEN D F, HU Z B. Thermal expansion properties of A2(MO4)3 (A=Ho and Tm; M=W and Mo) [J]. Solid State Sci, 2008, 10: 321-325.
[8] SUMITHRA S, TYAGI A K, UMARJI A M. Negative thermal expansion in Er2W3O12 and Yb2W3O12 by high temperature X-ray diffraction [J]. Mater Sci Eng B, 2005, 116: 14-18.
[9] SUMITHRA S, UMARJI A M. Negative thermal expansion in rare earth molybdates [J]. Solid State Sci, 2006, 8: 1453-1458.
[10] FORSTER P M, YOKOCHI A, SLEIGHT A W. Enhanced negative thermal expansion in Lu2W3O12 [J]. J Solid State Chem, 1998, 140: 157-158.
[11] SHANNON R D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides [J]. Acta Cryst, 1976, A32: 751-767.
[12] JIA Y Q. Crystal radii and effective ionic radii of the rare earth ions [J]. J Solid State Chem, 1991, 95: 184-187.
[13] SUMITHRA S, UMARJI A M. Role of crystal structure on the thermal expansion of Ln2W3O12 (Ln=La, Nd, Dy, Y, Er and Yb) [J]. Solid State Sci, 2004, 6: 1313-1319.
[14] ZHAO Xin-hua. The synthesis and structure of Al2Mo3-xWxO12 [J]. Chemical Journal of Chinese Universities, 1999, 20(3): 339-343. (in Chinese)
[15] SHEN Rong, WANG Tian-min. The synthesis and thermal expansion of Al2Mo3-xWxO12 [J]. Rare Metal Mater Eng, 2004, 33(1): 91-95. (in Chinese)
[16] RODRIGUEZ-CARVAJAL J. Program Fullprof.2k, version 3.30, Laboratoire Leon Brillouin[Z]. France, June 2005.
[17] LUBARDA V A. On the effective lattice parameter of binary alloys [J]. Mech Mater, 2003, 35: 53-68.
[18] LAMBREGTS M J, FRANK S. Application of Vegard’s law to mixed cation sodalites: A simple method for determining the stoichiometry [J]. Talanta, 2004, 62: 627-630.
[19] DUAN N, KAMESWARI U, SLEIGHT A W. Further contraction of ZrW2O8 [J]. J Am Chem Soc, 1999, 121: 10432-10433.
[20] XING X R, CHEN J, DENG J X, LIU G R. Solid solution Pb1-xSrxTiO3 and its thermal expansion [J]. J Alloys Compd, 2003, 360: 286-289.
Foundation item: Project (20080430556) supports by China Postdoctoral Science Foundation
Corresponding author: PENG Jie; Tel: +86-10-88236181; E-mail: pengjie@mails.gucas.ac.cn
DOI: 10.1016/S1003-6326(09)60079-0
(Edited by YANG Hua)

