
Effects of Na-substitution on structural and electronic properties of Li2CoSiO4 cathode material
WU Shun-qing(吴顺情)1, ZHU Zi-zhong(朱梓忠)1, YANG Yong(杨 勇)2, HOU Zhu-feng(侯柱锋)3
1. Department of Physics, Institute of Theoretical Physics and Astrophysics, Xiamen University,
Xiamen 361005, China;
2. State Key Lab for Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen 361005, China;
3. Department of Physics, Fudan University, Shanghai 200433, China
Received 16 September 2008; accepted 14 October 2008
Abstract: Na-substituted dilithium orthosilicate Li2CoSiO4 was investigated by performing density functional theory calculations within the GGA+U framework. The effects of Na-substitution on the electronic structures and structural properties of Li2CoSiO4 were presented. The results show that the Na-substitution on Li sites in Li2CoSiO4 induces a lowering of the conduction bands and a narrowing of the band gap, which could be helpful for enhancing the electronic conductivity. On the other hand, the Na-substitution on the Li ions in Li2CoSiO4 leads to the expansion of interlayer space of the adjacent corrugated layers. This lattice expansion effect would benefit the Li ion diffusion.
Key words: dilithium cobalt orthosilicate; Na-substitution; structural properties; electronic structures
1 Introduction
Polyanionic cathodes such as olivine type LiMPO4 (M=Mn, Fe, Co, Ni, etc)[1-3] have attracted considerable attention as next-generation cathodes due to their better safety characteristics compared with the more commonly used metal oxides, which act as strong oxidizers in charged state in direct contact with an organic electrolyte [4]. More recently, an entirely new class of lithium intercalation compounds based on silicates, Li2MSiO4, where M=Mn, Fe, Mn/Fe, Co, Ni, has been described [5-15]. Compared with phosphate materials in which only one lithium ion can be reversibly cycled, dilithium orthosilicate materials, i.e. Li2MSiO4, would allow reversible extraction of two lithium ions in principle, thus they should deliver higher capacity (e.g. Li2CoSiO4, 325 mA?h/g) than phosphates. This makes them very attractive new cathode materials for lithium ion batteries. Among these compounds, the dilithium cobalt orthosilicate (i.e. Li2CoSiO4) has been prepared successfully by a solution route and hydrothermal reaction[9]. However, the pristine Li2CoSiO4 prepared by solution route shows poor electrochemical performance due to its poor conductivity[9]. Even coated with carbon, reversible electrochemical extraction was limited to 0.46 Li per formula unit[9]. Further studies are essential to seek possible approaches to improve the electrical conductivity and the electrochemical performance. In the present work, we focus on the analysis of Na-substituted Li2CoSiO4, in order to characterize the structural properties and electronic structures of Li2-xNaxCoSiO4 (0<x<2), and to understand the effects induced by the Na-substitution on the related properties, including the electrical conductivity and lithium ion diffusion.
2 Computational details
All calculations on Li2-xNaxCoSiO4 were carried out by using the projector augmented wave(PAW) method [16] within the density functional theory(DFT) as implemented in the Vienna ab initio simulation package (VASP)[17-18]. The exchange and correlation energy was treated within the spin-polarized generalized gradient approximation(GGA) and parameterized by Perdew- Burke-Ernzerhof formula (PBE)[19]. Furthermore, the effects due to the localization of the d electrons of the transition metal ions in the silicates were taken into account with the GGA+U approach of DUDAREV et al[20]. Within this approach, an effective value of Ueff = (U-J)=6 eV was used for Li2-xNaxCoSiO4. Such a value was recently used to analyze the electronic structures and voltage of the Li2MSiO4[15]. The GGA+U approach has been proved to be able to well determine the structural and electronic properties of the LixMSiO4 (M=Mn, Fe, Co, Ni) compounds[15]. Wave functions were expanded in plane waves up to a kinetic energy cut-off of 500 eV. Brillouin-zone integrations were approximated by using special k-point sampling of Monkhorst-Pack scheme[21] with a 9×9×9 grid.
3 Results and discussion
The unit cell of Li2CoSiO4 has an orthorhombic structure in the Pmn21 space group with corrugated layers of SiO4 and CoO4 tetrahedra lying on the ac-plane and linked along the b-axis by LiO4 tetrahedra. This structure can also be described by a ZnS-type arrangement (in a distorted form of hexagonal close packing), within which the Zn sites are occupied by Li, Co and Si cations while the S sites are occupied by O anions. All the tetrahedra point in the same direction, perpendicular to the close-packed planes, and share only corners with each other[10]. Especially, Li ions occupy tetrahedral sites located between the two [SiO4-CoO4] corrugated layers (Fig.1). A path for lithium extraction/ insertion exists in this structure since the LiO4 tetrahedron is arranged in rows running along the a-axis by sharing corner. Also, the diffusion of Li ions in the corrugated layers is possible. There are two formula units per unit cell of Li2CoSiO4, i.e., each unit cell contains 4 lithium, 2 cobalt, 2 silicon, and 8 oxygen atoms. The structural stabilities, electronic structures and Li deintercalation for the dilithium orthosilicate Li2CoSiO4 have been investigated in our previous work[15].
For the Na-substituted compounds, i.e. Li2-xNaxCoSiO4, the initial crystal structures were obtained via replacing Li by Na in the corresponding optimized Li2CoSiO4 unit cells, then the crystallographic cell parameters (the cell shape as well as the lattice constants) and the atomic fractional coordinates were relaxed until the forces on all atoms were less than 0.01 eV/?. The calculated lattice parameters, equilibrium volumes for Li2-xNaxCoSiO4, with x=0, 0.5, 1 and 2, are listed in Table 1. To be more precise, the expansions of lattice parameter due to Na-substitution are also shown in Fig.2. The lattice parameters and equilibrium volume of Li2-xNaxCoSiO4 are well described by the VEGARD’s law[22] as shown in Fig.2. It is obvious that smaller x (the smaller amount of Na-substitution) would have less influence on the structural properties. This tendency also applies to the electronic structures, which will be discussed later. Furthermore, under the aim to improve the electrical conductivity of Li2CoSiO4, the predominance of the theoretical capacity is expected to be kept. Therefore, in the following we would focus mainly on the case of Li1.5Na0.5CoSiO4 and the comparison between the substituted Li1.5Na0.5CoSiO4 and the pure Li2CoSiO4. For the stable structures of Li1.5Na0.5CoSiO4, small deviations from the perfect orthogonality have been found, as listed in Table 1. Moreover, all the three lattice parameters of Li1.5Na0.5CoSiO4 are larger than those of Li2CoSiO4. This can be understood from the fact that the ionic radius in four-coordination of Na (0.99 ?) is larger than that of Li (0.59 ?)[23]. With respect to those of Li2CoSiO4, the deviations of the lattice parameters of Li1.5Na0.5CoSiO4 are 0.6% and 1.7% for a and c, respectively. Whereas, a more significant deviation (3.2%) has been found for the parameter b. The optimized cell volume is found to increase by about 5.5% from Li2CoSiO4 to Li1.5Na0.5CoSiO4.
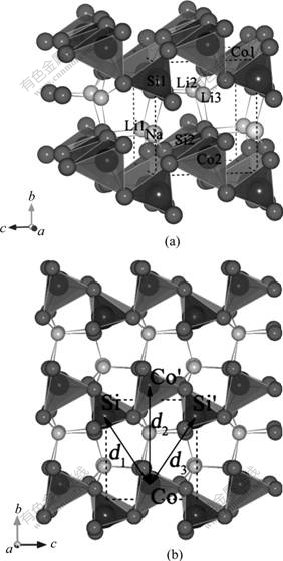
Fig.1 Structure of Li2-xNaxCoSiO4 (CoO4 tetrahedra: light gray; SiO4 tetrahedra: dark gray; Li/Na atoms: light (smaller/larger); O atoms: dark)
Table 1 Lattice parameters and equilibrium volumes for Li2-xNaxCoSiO4 (x =0, 0.5, 1, 2)
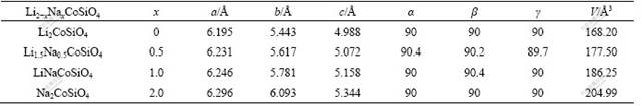
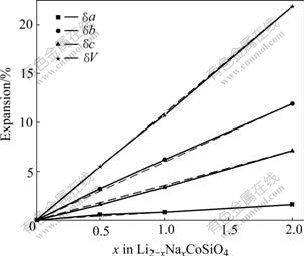
Fig.2 Expansion of lattice parameters and volumes due to Na-substitution compared with VEGARD’s prediction (dashed lines)[22]
On the other hand, as mentioned above, the corrugated layers should be two-dimensional(2D) pathways for lithium ion diffusion. Therefore, the interlayer space of the corrugated layers should be an important factor for the Li ion diffusion. According to the symmetry, three distances d1, d2 and d3 as shown in Fig.1(b) can be used to characterize the interlayer space of the corrugated layers. Table 2 lists the distances d1, d2 and d3 for Li2CoSiO4 and Li1.5Na0.5CoSiO4. It is obvious that all the three distances, i.e. the interlayer spaces, are enlarged when partial Li is replaced by Na. This means that the expansion of structural parameters leads to the expansion of interlayer space of the adjacent corrugated layers. This would benefit the lithium ion diffusion and may also further improve the electrochemical performance.
Another way of quantifying the structural differences is in terms of the bond lengths. The bond lengths of Co―O, Si―O, Li―O and Na―O for Li2CoSiO4 and Li1.5Na0.5CoSiO4 are listed in Table 3. Both the average bond lengths of Co―O and Si―O for Li1.5Na0.5CoSiO4 are almost the same as those for Li2CoSiO4, with a tiny increase of 0.3% and 0.1%, respectively. However, larger spread has been found for the Co―O bond lengths in Li1.5Na0.5CoSiO4, whereas the bond lengths of Si―O still keep a small spread. Although Na-substitution has an influence on the local environment, the Si―O bonds in Li1.5Na0.5CoSiO4 should still be strong covalent and afford the same lattice stabilization effect as in the Li2CoSiO4[14-15]. Besides, we can find that the average Li―O bond length of Na-substituted Li2CoSiO4 is slightly larger (1.5%) than that of the pure material. This indicates a wider Li ion pathway and lower Li-O interactions in Li1.5Na0.5CoSiO4.
In order to understand the effects of Na-substitution on the electronic structures, we calculated the band structures (Fig.3) and the density of states (DOS) (Fig.4) for the Li1.5Na0.5CoSiO4 and the pure Li2CoSiO4. In Fig.3, we can observe that the valence band maximum (VBM) and conduction band minimum (CBM) both occur at Γ-point, making both compounds to be direct gap materials. The gap value is about 3.74 eV for the pure Li2CoSiO4, and a smaller one (3.39 eV) for Li1.5Na0.5CoSiO4. This indicates that the Li1.5Na0.5CoSiO4 material has better electronic conductivity than the Li2CoSiO4 material. It is found that the Na-substitution induces the CBM’s shifting toward the Fermi level, especially for the Co projected-DOS (Fig.4(b)). The Li/Na, Si and O also contribute to the CBM and their CBM is also found to shift down, but the magnitude of their corresponding density of states is quite small compared with Co projected-DOS as shown in Figs.4(b)-(e). This means that the shifting of Co conduction bands would play an important role in the improvement of electronic conductivity when partial Li is substituted by Na. In addition, as shown in Fig.4(b), the Na-substitution has more significant influence on the Co2 atom, which has a smaller distance from the substituted Na than the Co1. The lowering of conduction band may be due to the expansion of the lattice and the distortion of the local environment (especially the CoO4 tetrahedra) caused by Na-substitution
Table 2 Distances d1, d2 and d3 as described in Fig.1(b) for Li2-xNaxCoSiO4 (x=0, 0.5)
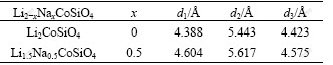
Table 3 Bond lengths for Li2-xNaxCoSiO4 (x=0, 0.5) (?)
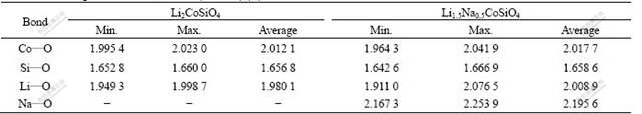
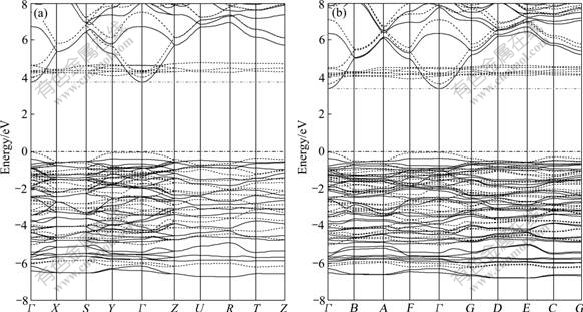
Fig.3 Band structure of Li2CoSiO4 (a) and Li1.5Na0.5CoSiO4 (b) (Solid lines are for majority spin and dash lines for minority spin. Points are labeled as Γ=(0, 0, 0), X(B)=(1/2, 0, 0), S(A)=(1/2, 1/2, 0), Y(F)=(0, 1/2, 0), Z(G)=(0, 0, 1/2), U(D)=(1/2, 0, 1/2), R(E)=(1/2, 1/2, 1/2), and T(C)=(0, 1/2, 1/2) with respect to (b1, b2, b3) for orthorhombic structure (a) and triclinic structure (b). Dash dot lines mark Fermi level (top of valence band))
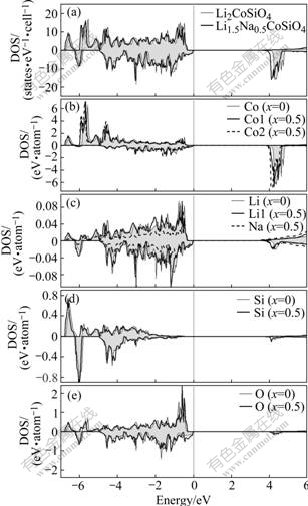
Fig.4 Total and projected density of states (PDOS) for Li2-xNaxCoSiO4 (x=0, 0.5): (a) Total DOS; (b) Co-PDOS; (c) Li/Na-PDOS; (d) Si-PDOS; (e) O-PDOS
.4 Conclusions
1) The effects of Na-substitution on the electronic structures and structural properties of Li2CoSiO4 are presented. When Li ions in Li2CoSiO4 are substituted (fully or partially) by Na ions, the lattice parameters expand especially for the parameter b. This lattice expansion effect benefits the Li ion diffusion due to the widening of the diffusion pathways.
2) The Na-substitution on Li sites in Li2CoSiO4 induces a lowering of the conduction bands and a narrowing of the band gap, which could be helpful for enhancing the electronic conductivity.
References
[1] PADHI A K, NANJUNDASWAMY K S, GOODENOUGH J B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries [J]. Journal of The Electrochemical Society, 1997, 144(4): 1188-1194.
[2] AMINE K, YASUDA H, YAMACHI M. Olivine LiCoPO4 as 4.8 V electrode material for lithium batteries [J]. Electrochemical and Solid-State Letters, 2000, 3(4): 178-179.
[3] LI G H, AZUMA H, TOHDA M. LiMnPO4 as the cathode for lithium batteries [J]. Electrochemical and Solid-State Letters, 2002, 5(6): A135-A137.
[4] ZHANG Z R, GONG Z L, YANG Y. Electrochemical performance and surface properties of bare and TiO2-coated cathode materials in lithium-ion batteries [J]. Journal of Physical Chemistry B, 2004, 108: 17546-17552.
[5] NYTEN A, ABOUIMRANE A, ARMAND M, GUSTAFSSON T, THOMAS J O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material [J]. Electrochemistry Communications, 2005, 7: 156-160.
[6] DOMINKO R, BELE M, GABERSCEK M, MEDEN A, REMSKAR M, JAMNIK J. Structure and electrochemical performance of Li2MnSiO4 and Li2FeSiO4 as potential Li-battery cathode materials [J]. Electrochemistry Communications, 2006, 8: 217-222.
[7] GONG Z L, LI Y X, YANG Y. Synthesis and characterization of Li2MnxFe1-xSiO4 as a cathode material for lithium-ion batteries [J]. Electrochemical and Solid-State Letters, 2006, 9(12): A542-A544.
[8] LI Y X, GONG Z L, YANG Y. Synthesis and characterization of Li2MnSiO4/C nanocomposite cathode material for lithium ion batteries [J]. Journal of Power Sources, 2007, 174: 528-532.
[9] GONG Z L, LI Y X, YANG Y. Synthesis and electrochemical performance of Li2CoSiO4 as cathode material for lithium ion batteries [J]. Journal of Power Sources, 2007, 174: 524-527.
[10] LYNESS C, DELOBEL B, ARMSTRONG A R, BRUCE P G. The lithium intercalation compound Li2CoSiO4 and its behaviour as a positive electrode for lithium batteries [J]. Chemical Communications, 2007: 4890-4892.
[11] GONG Z L, LI Y X, HE G N, LI J, YANG Y. Nanostructured Li2FeSiO4 electrode material synthesized through hydrothermal- assisted sol-gel process [J]. Electrochemical and Solid-State Letters, 2008, 11(5): A60-A63.
[12] LARSSON P, AHUJA R, NYTEN A, THOMAS J O. An ab initio study of the Li-ion battery cathode material Li2FeSiO4 [J]. Electrochemistry Communications, 2006, 8: 797-800.
[13] ARROYO-DE DOMPABLO M E, ARMAND M, TARASCON J M, AMADOR U. On-demand design of polyoxianionic cathode materials based on electronegativity correlations: An exploration of the Li2MSiO4 system (M=Fe, Mn, Co, Ni) [J]. Electrochemistry Communications, 2006, 8: 1292-1298.
[14] WU S Q, ZHANG J H, ZHU Z Z, YANG Y. Structural and electronic properties of the Li-ion battery cathode material LixCoSiO4 [J]. Current Applied Physics, 2007, 7: 611-616.
[15] WU S Q, ZHU Z Z, YANG Y, HOU Z F. Structural stabilities, electronic structures and lithium deintercalation in LixMSiO4 (M=Mn, Fe, Co, Ni): A GGA and GGA + U study [EB/OL]. Computational Materials Science, doi:10.1016/j.commatsci.2008.08.014.
[16] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Physical Review B, 1999, 59(3): 1758-1775.
[17] KRESSE G, FURTHMULLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Physical Review B, 1996, 54(16): 11169-11186.
[18] KRESSE G, FURTHMULLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Computational Materials Science, 1996, 6: 15-50.
[19] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple [J]. Physical Review Letters, 1996, 77(18): 3865-3868.
[20] DUDAREV S L, BOTTON G A, SAVRASOV S Y, HUMPHREYS C J, SUTTON A P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study [J]. Physical Review B, 1998, 57(3): 1505-1509.
[21] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188-5192.
[22] VEGARD L. The constitution of the mixed crystals and the filling of space of the atoms [J]. Zeitschrift für Physik, 1921, 5(1): 17-26.
[23] SHANNON R D. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides [J]. Acta Crystallographica, 1976, A32: 751-767.
Foundation item: Project(2007CB209702) supported by the National Basic Research Program of China; Projects(10774124, 20473068) supported by the National Natural Science Foundation of China
Corresponding author: ZHU Zi-zhong; Tel: +86-592-2182248; Fax: +86-592-2189426; E-mail: zzhu@xmu.edu.cn
DOI: 10.1016/S1003-6326(08)60249-6
(Edited by LI Xiang-qun)

