ОДХВ±ағЕӘғ1004-0609(2017)-01-0145-10
өУµНЖ·О»УЛОІүуЦРСх»ҮҢюіцУЛ
Ао ГЬ1, 2Ә¬ХЕ ±л1Ә¬ХЕПюОД1, 2Ә¬»Ж жғ1Ә¬¶ҰµВЬ°3Ә¬Т¶УВңь3
(1. ДП»ҒөуС§ »·ңі±Ә»¤Ул°ІИ«№¤іМС§ФғӘ¬ғвСф 421001Ә»
2. ДП»ҒөуС§ ·ЕЙдРФИэ·Пө¦АнУлө¦ЦГЦШµгКµСйКТӘ¬ғвСф421001Ә»
3. ДП»ҒөуС§ УЛүуТ±ЙъОпәәКх№ъ·АЦШµгС§үЖКµСйКТӘ¬ғвСф 421001)
ХҒ ТҒӘғХл¶ФµНЖ·О»УЛОІүуТтВцКҮғ¬БүёЯҰұОпПаёіөжЧөМ¬ёөФУ¶шФміЙµДУЛҢюіцВКµНµДОКМвӘ¬МбіцМнәУёЁЦъСх»ҮәБЖЖ»µВцКҮҢб№№¶шКµПЦЗү»ҮҢюіцУЛµДЛәВ·ҰӘІЙУГµӨТтЛШКµСй·Ё¶Ф±ИіӘ№жЛбҢюғН3ЦЦСх»ҮәБ(H2O2ҰұMnO2 ғНFe3+)Зү»ҮЛбҢю¶ФУЛҢюіцВКµДУ°ПмҰӘҢб№ы±нГчӘғµ±ҢюіцОВ¶ИҰұБтЛбЕЁ¶ИғНТғ№М±И·Ц±рОҒ30 ҰжҰұ1 mol/LғН20:1К±Ә¬ІЙУГіӘ№жЛбҢю6 hғуУЛµДҢюіцВКҢцОҒ78%Ә¬¶шФЪПаН¬µДҢюіцМхәюПВӘ¬Зү»ҮЛбҢю1.5 hУЛµДҢюіцВКүЙөпµҢ95%ҰӘҢюіцФьµДXRDә°SEM-EDS·ЦОцҢб№ы±нГчӘ¬H2O2ә°MnO2ңщДЬЖЖ»µВцКҮң§МеҢб№№Ә¬әхЙЩүЕБӘНЕңЫӘ¬µ«МнәУMnO2ғуЙъіЙРВµД№иЛбГМСОң§МеӘ¬Fe3+І»ДЬЖЖ»µВцКҮҢб№№Ә¬µ«ЖдСх»ҮЧчУГФЪТ»¶ЁіМ¶ИЙПДЬәУүмУЛµДҢюіцҰӘ
№ШәьөКӘғУЛОІүуӘ»ҢюіцӘ»Сх»ҮӘ»ВцКҮ
ЦРНә·ЦАағЕӘғTD983ҰҰҰҰ ОДПЧ±кЦңВлӘғA
ЛжЧЕғ˵繤ҵµДІ»¶П·ұХ№Ә¬ОТ№ъ¶ФУЛµДРиЗуБүҢ«іЦРшФцәУӘ¬Ф¤әЖµҢ2020ДкӘ¬ОҒВъЧгғЛµз·ұХ№ЛщРиТҒµДУЛҢ«өпµҢ6000 tЧуУТ[1]ҰӘДүЗ°Ә¬ОТ№ъУЛүуүҒІЙ·Ң·ЁЦчТҒУРіӘ№жҢБ°иҢюіцҰұ¶СҢюҰұФµШ±¬ЖЖҢюіцТФә°CO2µШҢюµИ[2]ҰӘФЪФµШ±¬ЖЖҢюіцТФә°CO2µШҢюµИ№¤ТХ±»үҒ·ұіцАөЦ®З°Ә¬іӘ№жҢБ°иҢюіцә°¶СҢюТ»Ц±КЗОТ№ъУЛүуүҒІЙµДЦчТҒ·Ң·ЁҰӘИ»¶шӘ¬ёГ№¤ТХИөІ»үЙ±ЬГвµШІъЙъБЛөуБүУЛОІүуҰӘңЭ№АәЖӘ¬ОТ№ъУЛОІүу¶СөжБүТСөпµҢ240ТЪt[3]ҰӘТтУЛОІүуЦРғ¬УР0.005%~0.03%µДУЛӘ¬ңҰ№ЬУЛЖҢңщЖ·О»ә«µНӘ¬µ«Ждғ¬УЛЧЬБүИөІ»үЙғцКУӘ¬і¤ЖЪ¶СөжІ»Ңц»б¶ФЦЬ±Я»·ңіФміЙСПЦШµД·ЕЙдРФОЫИң[4-5]Ә¬ТІ»бФміЙУЛЧКФөµДСПЦШАЛ·С[6]ҰӘ
УЙУЪОТ№ъУЛФүуңЯУРУЛЖ·О»µНҰұ°йЙъФҒЛШ¶аҰұүуРФёөФУµДМШµгӘ¬ОҒБЛМбёЯУЛүуµДҢюіцВКӘ¬ңҰ№ЬТСңүҒ·ұіцЕЁЛбКм»ҮёЯМъБЬВЛ¶СҢюәәКхҰұµНЙшНёРФүуКҮЦЖБӘ¶СҢюәәКхҰұПёБӘә¶үуКҮ¶СҢюәәКхҰұө®БҒ¶СҢюәәКхµИТ»ПµБРµДУЛүуЗү»ҮҢюіцКЦ¶О[7-9]Ә¬µ«УЙУЪУЛОпПаµДөжФЪРОМ¬ҰұУЛФЪВцКҮүуОпЦРµДёіөжЧөМ¬ҰұҢюіц¶ҮБ¦С§µИёчЦЦТтЛШµДУ°ПмӘ¬µәЦВУЛФүуµДҢюіцОІЙ°ЦРіӘІРөжУРТ»¶Ёғ¬БүµДУЛҰӘУЙУЪХвІү·ЦУЛТСңКЗФЪЗү»ҮҢюіцғуІРБфПВАөµДӘ¬өУ¶шµәЦВХвІү·ЦУЛІЙУГіӘ№жµДҢюіц·Ң·ЁДСТФҢшТ»ІҢ»ШКХӘ¬µ«ңНДүЗ°ОТ№ъµДУЛЧКФөөұБүПЦЧө¶шСФӘ¬ёГОІүуүЙүөЧчКЗТ»ЦЦә«Ж¶ДСТ±УЛүуЧКФөҰӘ
№ъДЪНвХл¶Фә«Ж¶ДСТ±УЛүуЧКФөЦРУЛµДҢюіцүҒХ№БЛөуБүµДСРңүӘ¬ёЕАЁЖрАөЦчТҒУРБҢЦЦЛәВ·ӘғЖдЦРЧоіӘУГµД·ҢКҢКЗНЁ№эёД±дҢюіцәБµДЧйіЙАөКµПЦУЛµДёЯР§ҢюіцӘ¬ИзHClҢюіцҰұHNO3ҢюіцҰұМәЛбСОҢюіцµИ[10-11]Ә¬»тХЯМнәУёчЦЦСх»ҮәБИзO2ҰұNa2O2ҰұFe3+µИҢ«ДСИЬµДЛДәЫУЛЧҒ»ҮОҒТЧИЬµДU6+[12-14]ҰӘЖдөОКЗІЙУГНвіҰЗү»ҮКЦ¶ОМбёЯУЛµДҢюіцВКӘ¬LEIµИ[15]СРңүБЛВИ»ҮёЖ±ғЙХ-ПхЛбҢюіцБҢ¶О№¤ТХөУ·ЫГғ»ТЦР»ШКХУЛӘ¬LADOLAµИ[16]МҢМЦБЛі¬ЙщІЁёЁЦъ¶ФУЛҢюіцµДУ°ПмӘ¬СоУкЙҢµИ[17]ІЙУГОұІЁәУИИФ¤ө¦Ан¶СҢюУЛүуКҮАөёДЙЖУЛµДҢюіцМхәюҰӘ И»¶шӘ¬УЙУЪә«Ж¶УЛүуКҮЦчТҒіЙ·ЦОҒ№иЛбСОВцКҮӘ¬ЖдЦРУЛғ¬Бүә«µНӘ¬ЗТТФОұПёБӘµДРОКҢ·ЦЙұУЪВцКҮүуОпЦРҰӘТтөЛӘ¬ИзғОЖЖ»µВцКҮҢб№№¶шКµПЦУЛµДҢвАлүЙДЬКЗМбёЯУЛҢюіцВКµДБнТ»ЦЦРВЛәВ·ҰӘ
±ңОДЧчХЯТФµНЖ·О»УЛОІүуОҒСРңү¶ФПуӘ¬ДвНЁ№эіӘ№жЛбҢюә°Сх»ҮҢюіцµД¶Ф±ИСРңүӘ¬МҢМЦВцКҮ-ИЬҢюәБЦ®әдµДОұ·өУ¦¶ФУЛҢюіцВКµДУ°ПмӘ¬үәІмБЛH2O2ҰұMnO2ғНFe3+ЧчОҒСх»ҮәБК±УЛОІүуЦРµДВцКҮФЪҢюіц№эіМЦРµДОұ№ЫҢб№№µД±д»ҮҰӘ
1 КµСй
1.1 КµСйФБП
±ңСРңүЦРЛщУГУЛОІүуАөЧФУЪғюДПДіУЛОІүуүвӘ¬ёГУЛОІүуЦчТҒІъЙъУЪө«НіµДУЛүу¶СҢю№¤ТХӘ¬СщЖ·ИҰ»ШғуКЧПИңЛД·Ц·Ёід·Ц»мФИӘ¬И»ғуФЪ105 ҰжµДғжПдЦР±ӘіЦ24 hӘ¬өэЧФИ»АдИөғуӘ¬ИҰЙЩБүФОІүуУГУЪБӘң¶·Цә¶ә°РОГІ·ЦОцӘ¬ЖдУаОІүуІЙУГХс¶ҮДӨСщ»ъДӨПёЦБТ»¶ЁБӘ¶ИӘ¬өэҢшРРБӘң¶·Цә¶ғу±ӘөжУЪёЙФпЖчГуЦРӘ¬ТФ±ёғуРшКµСйК№УГҰӘ
1.2 ЦчТҒКµСйЙи±ё
Хс¶ҮДӨСщ»ъ(ZM-200BРН)Ә»ЕД»чКҢ±кЧәХсЙё»ъ(XSBP-AРН)Ә»ОұүШКэПФµзИИ°е(EH45BРН)Ә»ғгОВЛ®ФҰТҰөІ(SWB-2000РН)Ә»µзЧУМмЖҢ(BSA224SРН)Ә»µзИИ№Д·зёЙФпПд(HG101-4AРН)Ә»XЙдПЯСЬЙдТЗ(Rigaku Ultima-IVРН)Ә»ЙЁГиµзңµ(Hitach S4800РН)Ә»µзёРсоғПµИАлЧУМеФЧУ·ұЙд№вЖЧТЗ(PS-6ХжүХРН)ҰӘ
1.3 КµСйФАн
Т»°гУЛОІүуЦРЦчТҒөжФЪU4+УлU6+БҢЦЦәЫМ¬Ә¬¶шҢ«ДСИЬµДЛДәЫУЛЧҒ»ҮОҒТЧИЬµДU6+КЗМбёЯУЛҢюіцВКµДЗ°МбҰӘіӘ№жЛбҢю№эіМЦРОөМнәУИОғОСх»ҮәБК±Ә¬ФтЦчТҒАыУГүХЖшЦРµДСхЧчОҒСх»ҮәБӘ¬ИфҢюіц№эіМЦРМнәУСх»ҮәБӘ¬ФтДЬәУЛЩУЛµДСх»ҮӘ¬Ңюіц№эіМЦРүЙДЬ·ұЙъµД»ҮС§·өУ¦ИзПВЛщКңӘғ
2UO2+O2Ұъ2UO3 (1)
UO3+2H+ҰъUO22++H2O (2)
UO22++nSO42-ҰъUO2(SO4)n2-2n (3)
UO2+H2O2ҰъUO3+H2O (4)
UO2+2Fe3+ҰъUO22+ +2Fe2+ (5)
4Fe2++O2+4H+Ұъ4Fe3++2H2O (6)
2Fe2++MnO2+4H+Ұъ2Fe3++Mn2++2H2O (7)
1.4 КµСй·Ң·Ё
1) БӘң¶·Цә¶·Ң·Ё
ЧәИ·іЖИҰ100 gғжёЙғуµДУЛОІүуСщЖ·Ә¬·ЕИлТС°өХХЙёНшүЧң¶өУРҰµҢөуµДЛіРтЕЕБРµДЦ±ң¶200 mmµД±кЧәЙёЦР(ЙёНшүЧң¶425Ұұ250Ұұ150Ұұ74ғН55 ¦Мm)Ә¬Ң«±кЧәЙёЦГУЪЕД»чКҢ±кЧәХсЙё»ъЙПЙё·Ц30 minӘ¬·Ц±ріЖИҰГүёцДүКэПВУЛОІүуµДЦКБүӘ¬ІұН¬К±·ЦОцёГДүКэПВУЛОІүуЦРУЛµДЖ·О»ҰӘёщңЭКµСйҢб№ыӘ¬И·¶ЁЧоәСµДБӘң¶ғуӘ¬ІЙУГХс¶ҮДӨСщ»ъҢ«УЛОІүуҢшРРДӨПёӘ¬өэИ«ІүүЕБӘНЁ№эёГБӘң¶µД±кЧәЙёғуӘ¬Ң«ДӨПёµДСщЖ·ід·Ц»мФИғуУГУЪғуРшҢюіцКµСйСРңүҰӘ
2) Ңюіц·Ң·Ё
ЧәИ·іЖИҰТ»¶ЁБүµДУЛОІүуСщЖ·Ә¬ЦГУЪ250 mLµДҢфүЪЧ¶РОЖүЦРӘ¬°өХХТ»¶ЁµДТғ№М±ИМнәУФ¤ЙиЕЁ¶ИµДҢюіцТғә°Сх»ҮәБӘ¬ЗбЗбТҰ»ОғуФЪЧ¶РОЖүүЪёЗЙПНёЖшПрҢғИы·АЦ№Ңюіц№эіМЦРТғМеХф·ұӘ¬Ң«Ч¶РОЖүЦГУЪғгОВЛ®ФҰТҰөІЦРӘ¬ІЙУГ»ШРэХрµө·ҢКҢҢюіцӘ¬»ШРэЛЩ¶И±ӘіЦОҒ200 r/minӘ¬ҢюіцҢбКшғуІЙУГүмЛЩ¶ЁРФВЛЦҢ¶ФҢюіцТғҢшРР№эВЛӘ¬ИҰ№эВЛТғҢшРРУЛғ¬Бү·ЦОцӘ¬ІұәЖЛгУЛµДҢюіцВКӘ¬ҢюіцФьЦ±ҢУғжёЙғу±ӘБфӘ¬УГУЪXRDҰұSEMµИ·ЦОцҰӘ
3) ·ЦОц·Ң·Ё
УЛОІүуµД»ҮС§іЙ·ЦІЙУГµзёРсоғПµИАлЧУМеФЧУ·ұЙд№вЖЧТЗ·ЦОцӘ»УЛОІүуµДОпПаЧйіЙІЙУГXЙдПЯСЬЙдТЗҢшРР·ЦОцӘ»РОГІМШХчІЙУГЙЁГиµзңµҢшРР·ЦОцӘ»УЛОІүуә°ҢюіцТғЦРУЛµДғ¬БүІЙУГ·°Лб淋ζЁ·ЁҢшРР·ЦОцҰӘУЛОІүуЦРУЛµДҢюіцВКІЙУГКҢ(8)әЖЛгӘғ
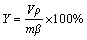 (8)
(8)
КҢЦРӘғYОҒУЛОІүуЦРУЛµДҢюіцВКӘ¬%Ә»VОҒҢюіцТғМе»эӘ¬LӘ» ОҒҢюіцТғЦРУЛµДЕЁ¶ИӘ¬g/LӘ»mОҒҢюіцЗ°УЛОІүуµДЦКБүӘ¬gӘ»¦ВОҒҢюіцЗ°УЛОІүуЦРУЛµДғ¬БүӘ¬%ҰӘ
ОҒҢюіцТғЦРУЛµДЕЁ¶ИӘ¬g/LӘ»mОҒҢюіцЗ°УЛОІүуµДЦКБүӘ¬gӘ»¦ВОҒҢюіцЗ°УЛОІүуЦРУЛµДғ¬БүӘ¬%ҰӘ
2 Ңб№ыУлМЦВЫ
2.1 УЛОІүуРФЦК·ЦОц
ІЙУГICP¶ФУЛОІүуµДµД»ҮС§ЧйіЙҢшРРБЛ·ЦОцӘ¬·ЦОцҢб№ыИз±н1ЛщБРҰӘУЛОІүуЦРУЛғ¬БүғЬµНӘ¬ФәОҒ0.008%(ЦКБү·ЦКэ)Ә¬КфУЪµНЖ·О»УЛОІүуӘ»МъФҒЛШғ¬БүёЯӘ¬ФЪҢюіц№эіМЦРүЙЖрµҢТ»¶ЁµДөЯ»ҮСх»ҮЧчУГӘ»ВБғ¬БүҢП¶аӘ¬¶шёЖҰұГңғ¬БүПа¶ФҢПЙЩӘ¬БнНвОІүуСщЦРғ¬УРТ»¶ЁµДБтФҒЛШӘ¬ФЪҢюіц№эіМЦРОҒБЛҢПЙЩЛбµДПығДӘ¬ТЛСҰУГБтЛбЧчОҒҢюіцәБҰӘ
УЛОІүуБӘң¶µД·ЦІәТФә°І»Н¬БӘң¶ПВУЛµДғ¬БүКЗңц¶ЁУЛОІүуЦРУЛҢюіцР§№ыµДЦчТҒТтЛШЦ®Т»Ә¬НЁ№э¶ФФКәУЛОІүуІ»Н¬БӘң¶ПВУЛµДғ¬Бү·ЦОцӘ¬үЙТФіхІҢЕР¶ПФУЛүуФЪҢюіц№эіМЦРРиТҒµДҢПККТЛµДБӘң¶·¶О§Ә¬өУ¶шОҒОІүуЗү»ҮҢюіцЦРБӘң¶µДүШЦЖМṩТАңЭҰӘТтөЛӘ¬±ңКµСйЦРІЙУГЙё·Ц·Ё·ЦОцБЛУЛОІүуµДБӘң¶·ЦІәӘ¬НЁ№э±кЧәЙёҢ«УЛОІүу·ЦіЙөуУЪ425Ұұ425~250Ұұ250~150Ұұ150~74Ұұ74~55ТФә°РҰУЪ55 ¦МmµД6ёцІ»Н¬БӘң¶µДСщЖ·Ә¬УЛОІүуµДБӘң¶·ЦІәВКә°УЛғ¬Бү·ЦОцҢб№ыИз±н2ЛщБРҰӘ
±н1 УЛОІүуµД ICP·ЦОцҢб№ы
Table 1 ICP results of uranium tailings

±н2 УЛОІүуБӘң¶Йё·ЦКµСйҢб№ы
Table 2 Results on particle size distribution of uranium tailings
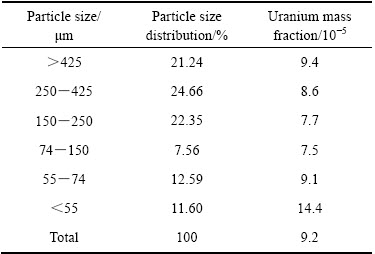
УЙ±н2үЙЦҒӘ¬УЛОІүуµДБӘң¶·ЦІә»щ±ңңщФИӘ¬µ«ЧЬМеүЕБӘБӘң¶Ж«өуӘ¬ЖдЦРБӘң¶өуУЪ150 ¦МmµДүЕБӘХәЧЬБүµД75%ТФЙПӘ¬ХвЦчТҒКЗУЙФүу¶СҢю№эіМЦРүЕБӘіЯөзЛщңц¶ЁµДҰӘөУёчБӘң¶·¶О§ДЪУЛғ¬БүµД·ЦОцҢб№ыүЙЦҒӘ¬УЛғ¬БүФЪІ»Н¬БӘң¶ПВµД·ЦІәІо±рҢПөуӘ¬ЖдЦР55 ¦МmТФПВµДРҰБӘң¶ЦРУЛғ¬БүЧоёЯӘ¬өпµҢ0.014%Ә¬ТСі¬№эУЛОІүуµДЖҢңщУЛғ¬Бү0.008%Ә¬ЛжЧЕүЕБӘБӘң¶µДФцөуӘ¬УЛғ¬БүТІЦрҢӨҢµµНӘ¬µ«БӘң¶і¬№э74 ¦МmТФЙПӘ¬УЛғ¬БүУЦүҒКәПФЦшФцәУҰӘФміЙУЛФЪІ»Н¬БӘң¶ПВµДғ¬БүІоТмЦчТҒУлФУЛүуµД¶СҢю№¤ТХУР№Ш[18]ҰӘ¶СҢю№эіМЦРИфүЕБӘҢПРҰӘ¬І»ҢцЧиЦНҢюіцТғµДЙшБчӘ¬µәЦВҢюіцТғОЮ·ЁУлүЕБӘЦРµДУЛОпПаҢУөӨӘ¬өУ¶шЦ±ҢУҢµµНБЛПёүЕБӘЦРУЛµДҢюіцР§№ы[19]Ә»іэөЛЦ®НвӘ¬БӘң¶ҢПРҰµДүЕБӘФЪҢюіц№эіМЦРИЭТЧІъЙъДа»ҮПЦПуӘ¬өУ¶ш¶ФҢюіцТғЦРµДУЛІъЙъДжОьёҢЧчУГӘ¬ФЪТ»¶ЁіМ¶ИЙПТІҢµµНБЛУЛµДҢюіцР§№ыӘ¬ЧоЦХ±нПЦОҒПёБӘң¶ә¶±рµДУЛОІүуЦРУЛғ¬БүЖ«ёЯӘ»Иф¶СҢю№эіМЦРУЛүуБӘң¶ҢПөуӘ¬УЙУЪУЛүуЦРУЛµДЖ·О»Т»°гҢПµНӘ¬¶СҢю№эіМЦР»бУ°ПмУЛФЪҢюіцТғЦРµД±©В¶ВКӘ¬µәЦВҢюіцОІүуЦРөуБӘң¶ә¶±рµДүЕБӘЦРУЛғ¬БүПФЦшФцәУҰӘЧЫғПТФЙП·ЦОцүЙЦҒӘ¬ОҒБЛ»сµГҢПёЯµДУЛҢюіцВКӘ¬±ШРлҢ«УЛОІүуµДБӘң¶үШЦЖФЪ74 ¦МmЧуУТµД·¶О§ДЪҰӘ
УЛОІүуµДXRDЖЧИзНә1ЛщКңҰӘУЛОІүуµДЦчТҒОпПаУРSiO2ҰұМъ№иЛбСО(Fe3FeSiO4(OH)5)ғНВБ№иЛбСО(BaAl2Si2O8)Ә¬µ«УЛµДғ¬Бүә«µНӘ¬Тт¶шОөәмІвµҢУЛµДОпПаҰӘУЙУЪУЛОІүуµДЦчТҒіЙ·ЦОҒВцКҮүуОпӘ¬ОҒБЛңҰүЙДЬМбёЯУЛµДҢюіцВКӘ¬±ШРлЖЖ»µВцКҮҢб№№Ә¬Ң«УЛОпПаөУВцКҮүуОпЦРҢвАліцАө[20]ҰӘТ»°г¶шСФӘ¬УЙУЪSiO2РФЦКОИ¶ЁӘ¬ФЪЛбРФМхәюПВДСТФЦ±ҢУЖЖ»µӘ¬ТтөЛӘ¬±ңСРңүЦРДвНЁ№эМнәУСх»ҮәБАөЖЖ»µМъ№иЛбСОә°ВБ№иЛбСОµИВцКҮҢб№№Ә¬ФЪТ»¶ЁіМ¶ИЙПҢ«УЛҢвАліцАөӘ¬өУ¶шМбёЯУЛµДҢюіцВКҰӘ

Нә1 УЛОІүуµДXRDЖЧ
Fig. 1 XRD pattern of uranium tailings
УЛОІүуµДРОГІә°ДЬЖЧ·ЦОцҢб№ыәыНә2ЛщКңҰӘУЛОІүуµДүЕБӘБӘң¶·ЦІәФЪ10~400 ¦МmЧуУТӘ¬іКІ»№жФтµДРОЧөӘ¬ФЪҢюіцЗ°үЙүәВЗІЙУГДӨүу·ЁәхРҰүЕБӘБӘң¶ҰӘЖдДЬЖЧ·ЦОцҢб№ы±нГчӘ¬УЛОІүуµДЦчТҒФҒЛШОҒSiҰұAlҰұFeӘ¬УлXRD·ЦОцҢб№ы»щ±ңТ»ЦВҰӘ

Нә2 УЛОІүуµДSEMПсә°EDS·ЦОцҢб№ы
Fig. 2 SEM image(a) and EDS results(b) of uranium tailings
2.2 УЛОІүуіӘ№жЛбҢюКµСйҢб№ы
1) БӘң¶¶ФУЛҢюіцµДУ°Пм
ФЪҢюіцОВ¶ИОҒ30 ҰжҰұЧҒЛЩ200 r/mҰұТғ№М±ИОҒ20:1Ә¬БтЛбЕЁ¶ИОҒ1 mol/LҰұҢюіцК±әдОҒ6 hµДМхәюПВӘ¬үәІмБӘ¶И¶ФУЛҢюіцВКµДУ°ПмӘ¬КµСйҢб№ыИзНә3ЛщКңҰӘөУНә3үЙТФүөіцӘ¬ЛжЧЕБӘң¶µДФцөуӘ¬УЛµДҢюіцВКЦрҢӨәхРҰҰӘµ±ҢюіцБӘң¶РҰУЪ55 ¦МmК±Ә¬УЛҢюіцВКОҒ80%Ә¬µ±БӘң¶ФЪ55~74 ¦Мm·¶О§ДЪК±Ә¬ҢюіцВКҢµµНЦБ73%Ә¬ҢшТ»ІҢФцөуБӘң¶Ә¬УЛҢюіцВКҢ«ПФЦшҢµµНҰӘңҰ№ЬБӘң¶ФЪ55 ¦МmТФПВК±үЙ»сµГҢПёЯµДҢюіцВКӘ¬µ«ОІүуөУ74 ¦МmДӨПёЦБ55 ¦МmЖдДӨүуК±әдә°ДЬғДҢ«ПФЦшФцәУӘ¬Н¬К±ТІүәВЗµҢҢюіцғуµД№эВЛ¶ВИыОКМвҰӘТтөЛӘ¬±ңСРңүЦРғуРшҢюіцКµСйңщҢ«ОІүуДӨПёЦБРҰУЪ74 ¦МmҰӘ

Нә3 БӘң¶¶ФУЛҢюіцВКµДУ°Пм
Fig. 3 Effects of particle size on uranium extraction ratio
2) БтЛбЕЁ¶И¶ФУЛҢюіцµДУ°Пм
ФЪҢюіцОВ¶ИОҒ30 ҰжҰұЧҒЛЩОҒ200 r/mҰұТғ№М±ИОҒ20:1ҰұҢюіцК±әдОҒ6 hµДМхәюПВӘ¬үәІмБтЛбЕЁ¶И¶ФУЛҢюіцµДУ°ПмҰӘ

Нә4 БтЛбЕЁ¶И¶ФУЛҢюіцВКµДУ°Пм
Fig. 4 Effects of H2SO4 concentration on uranium extraction ratio
УЙНә4үЙЦҒӘ¬БтЛбЕЁ¶ИµНУЪ0.5 mol/LК±Ә¬УЛµДҢюіцВКЛжБтЛбЕЁ¶ИµДФцөу¶шПФЦшФцәУӘ¬µ±БтЛбЕЁ¶Иі¬№э0.5 mol/LғуӘ¬БтЛбЕЁ¶ИµДФцәУ¶ФУЛҢюіцВКµДУ°ПмЦрҢӨҢµµНҰӘХвКЗУЙУЪФЪБтЛбЕЁ¶ИҢПµНµДЗйүцПВӘ¬µ±Тғ№М±ИТ»¶ЁК±Ә¬үуСщ±нГж·ұЙъ»ҮС§·өУ¦К±БтЛбБүө¦УЪ¶МИ±µДЧөМ¬Ә¬ТтөЛФцәУБтЛбµДЕЁ¶ИУРАыУЪУЛµДҢюіцӘ»¶шµ±БтЛбЕЁ¶ИөпµҢТ»¶ЁіМ¶ИК±Ә¬БтЛбө¦УЪ№эБүЧөМ¬Ә¬µ«УЙУЪБтЛбОЮ·ЁЖЖ»µОІүуЦРµДВцКҮҢб№№Ә¬µәЦВІү·ЦУЛОЮ·ЁҢвАлӘ¬ТтөЛәМРшФцәУБтЛбЕЁ¶И¶ФУЛҢюіцВКµДУ°ПмІ»өуҰӘ
3) Тғ№М±И¶ФУЛҢюіцµДУ°Пм
ФЪҢюіцОВ¶ИОҒ30 ҰжҰұЧҒЛЩОҒ200 r/mҰұБтЛбЕЁ¶ИОҒ1 mol/LҰұҢюіцК±әдОҒ6 hµДМхәюПВӘ¬үәІмТғ№М±И¶ФУЛҢюіцВКµДУ°ПмӘ¬КµСйҢб№ыИзНә5ЛщКңҰӘУЙНә5үЙЦҒӘ¬Тғ№М±И¶ФУЛµДҢюіцВКУРПФЦшУ°ПмӘ¬ЛжЧЕТғ№М±ИµДЦрҢӨФцөуӘ¬УЛµДҢюіцВКПФЦшМбёЯҰӘµ«Тғ№М±Иі¬№э20ТФғуӘ¬УЛµДҢюіцВКҢцөУ78%ФцәУЦБ82%Ә¬ТтөЛӘ¬ғуРшКµСйµДТғ№М±Иңщ№М¶ЁФЪ20ҰӘ

Нә5 Тғ№М±И¶ФУЛҢюіцВКµДУ°Пм
Fig. 5 Effects of liquid to solid ratio on uranium extraction ratio
4) ҢюіцК±әд¶ФУЛҢюіцµДУ°Пм
ФЪҢюіцОВ¶ИОҒ30 ҰжҰұЧҒЛЩОҒ200 r/mҰұБтЛбЕЁ¶ИОҒ1 mol/LҰұТғ№М±ИОҒ20µДМхәюПВӘ¬үәІмҢюіцК±әд¶ФУЛҢюіцВКµДУ°ПмӘ¬КµСйҢб№ыИзНә6ЛщКңҰӘөУНә6үЙТФүөіцӘ¬ФЪҢюіцК±әдОҒ1 hДЪӘ¬УЛµДҢюіцВКҢц45%ЧуУТӘ¬ёГҢЧ¶ОүЙДЬЦчТҒОҒБтЛб»ғВэЗЦКөВцКҮµД№эіМӘ»ЛжЧЕ·өУ¦К±әдµДСУі¤Ә¬ҢПТЧҢвАлµДІү·ЦУЛЦрҢӨөУВцКҮүуОпЦР°юАлӘ¬Тт¶шУЛµДҢюіцВКЦрҢӨМбёЯӘ»µ±К±әді¬№э4 hғуӘ¬УЛµДҢюіцВК±д»ҮІ»ГчПФӘ¬µ«ЧоёЯУЛҢюіцВКТІҢц78%ЧуУТӘ¬ЛµГчБЛУЛµДҢюіцКЬµҢБЛЧи°ҰӘ
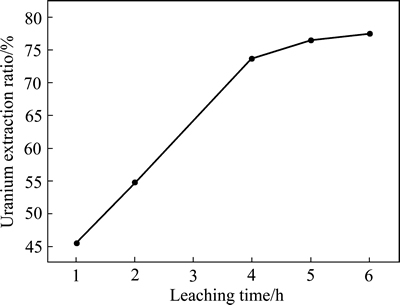
Нә6 ҢюіцК±әд¶ФУЛҢюіцВКµДУ°Пм
Fig. 6 Effects of reaction time on uranium extraction ratio
2.3 Сх»ҮҢюіцКµСйҢб№ы
ФЪіӘ№жЛбҢю№¤ТХЦРәөК№ҢюіцК±әді¤өп6 hӘ¬УЛµДҢюіцВКҢц78%ЧуУТӘ¬ОҒБЛМбёЯУЛµДҢюіцВКә°Лх¶МҢюіцК±әдӘ¬±ңКµСйЦРФЪіӘ№жЛбҢюµД»щөҰЙПӘ¬ДвНЁ№эЗү»ҮСх»ҮҢюіцµД·ҢКҢёДЙЖУЛµДҢюіцР§№ыӘ¬·Ц±рСҰИҰБЛ3ЦЦңЯУРөъ±нРФµДСх»ҮәБ(H2O2Ә¬MnO2ТФә°Fe2(SO)3)Ә¬ФЪҢюіцОВ¶ИОҒ30 ҰжҰұЧҒЛЩОҒ200 r/mҰұH2SO4ЕЁ¶ИОҒ1 mol/LғНТғ№М±ИОҒ20:1µДМхәюПВӘ¬·Ц±рүәІмСх»ҮәБµДМнәУБүә°ҢюіцК±әд¶ФУЛҢюіцВКµДУ°ПмӘ¬КµСйҢб№ыИзНә7ЛщКңҰӘ

Нә7 Сх»ҮәБМнәУБү¶ФУЛҢюіцВКµДУ°Пм
Fig. 7 Effects of oxidant concentration on uranium extraction ratio
УліӘ№жЛбҢю№¤ТХПа±ИӘ¬Сх»ҮәБ¶ФФцЗүУЛµДҢюіцВКңЯУРПФЦшµДР§№ыӘ¬ФЪ1.5 hДЪӘ¬УЛµДҢюіцВКәөүЙөпµҢ·еЦµӘ¬ЗТЧоөуУЛҢюіцВКүЙМбёЯЦБ95%ҰӘµ«Сх»ҮәБµДМнәУБү¶ФУЛµДҢюіцңЯУРҢПөуµДУ°ПмҰӘФЪСх»ҮәБМнәУБүҢПµНК±Ә¬MnO2µДЗү»ҮР§№ыЧоәСӘ¬µ«ЛжЧЕMnO2МнәУБүµДФцәУӘ¬ЖдЗү»ҮР§№ыГчПФПВҢµӘ¬ХвЦчТҒКЗТтОҒ¶юСх»ҮГМ±ңЙнКЗУЛµДБәғГОьёҢәБ[21-22]Ә¬ЙЩБүМнәУУРЦъУЪЖд¶ФВцКҮҢб№№µДЖЖ»µӘ¬№эБүМнәУҢ«µәЦВТСҢюіцµДУЛФЩөООьёҢµҢ¶юСх»ҮГМ±нГжӘ¬ФміЙУЛЧЬМеҢюіцВКµДҢµµНҰӘ¶ФУЪH2O2¶шСФӘ¬ЛжЧЕЖдМнәУБүµДФцәУӘ¬УЛµДҢюіцВКПФЦшФцәУӘ¬ЛµГчЖд¶ФөЩҢшУЛµДҢвАлңЯУР»эә«Р§№ыӘ¬ХвУлLASHEENµИµДКµСйҢб№ыТ»ЦВ[23]ҰӘFe3+¶ФУЛµДҢюіцОЮГчПФУ°ПмҰӘ
ҢюіцК±әд¶ФУЛµДҢюіцВКТІУРҢПөуµДУ°ПмӘ¬µ±Сх»ҮәБОҒH2O2ғНMnO2К±Ә¬ФЪ·өУ¦үҒКәµД1.5 hДЪӘ¬УЛµДҢюіцВКүмЛЩМбёЯӘ¬µ±·өУ¦К±әдФЪ1.5~3 hЦ®әдК±Ә¬УЛµДҢюіцВКңщУР·ЗіӘГчПФµДПВҢµЗчКЖӘ¬ХвүЙДЬКЗТтОҒH2O2ғНMnO2ФЪ·өУ¦іхКәҢЧ¶ОЦчТҒТФЖЖ»µ№иЛбСОВцКҮҢб№№ОҒЦчӘ¬өЩҢшБЛУЛµДҢвАлӘ¬µ«Н¬К±ВцКҮҢб№№µДЖЖ»µӘ¬±ШИ»µәЦВУЛОІүу±И±нГж»эә°үЧП¶ВКµДФцәУӘ¬өУ¶шәУЗүБЛВцКҮүуОп¶ФУЛµДОьёҢЧчУГӘ¬БнНвӘ¬Сх»ҮәБµДМнәУүЙДЬ»бөЩК№РВµД№иЛбСОүуОпµДІъЙъӘ¬¶ш№иЛбСОүуОпТІКЗУЛµДБәғГОьёҢәБ[24]ҰӘТтөЛӘ¬ФЪёГҢЧ¶ОУЛµДҢюіцВКіКПЦПВҢµЗчКЖҰӘµ±ҢюіцК±әдәМРшСУі¤К±Ә¬УЛµДҢюіцВКіКПЦ»ғВэЙПЙэ»тЗчУЪЖҢғвҰӘ¶шFe3+µДЗү»ҮЧчУГ№жВЙУлH2O2ғНMnO2УРГчПФІоТмҰӘµ±Fe3+МнәУБүҢПµНК±Ә¬Ңюіц№эіМЦРГ»УРіцПЦҢюіцВКПВҢµµДЗчКЖӘ¬ХвТІЛµГчFe3+µДСх»ҮЧчУГ»ъАнІұ·ЗЦ±ҢУЖЖ»µ№иЛбСОВцКҮҢб№№Ә¬¶шКЗҢ«УЛОІүуЦРДСИЬµДµНәЫУЛСх»ҮОҒёЯәЫУЛӘ¬ІЕ»бµәЦВFe3+µДСх»ҮР§№ыПФЦшµНУЪH2O2ғНMnO2ҰӘµ«Fe3+МнәУБүөпµҢ0.5 mol/LК±Ә¬·өУ¦2 hғуУЛµДҢюіцВКіКПЦүмЛЩПВҢµЗчКЖӘ¬ХвүЙДЬКЗТтОҒёЯЕЁ¶ИµДFe3+ФЪі¤К±әд·өУ¦ғуә«ТЧЙъіЙңЯУРёЯР§РхДэЧчУГµДңЫғПБтЛбМъ»тЗвСх»ҮМъҢғ Ме[25]Ә¬ЦВК№ҢюіцТғЦРµДУЛ±»ОьёҢµҢОІүуүЕБӘ±нГжҰӘ
2.4 Сх»ҮәБ¶ФУЛОІүуЗү»ҮҢюіцµДЧчУГ»ъАн·ЦОц
ОҒБЛҢшТ»ІҢІйГчСх»ҮәБФЪУЛОІүуҢюіц№эіМЦРµДЧчУГ»ъАнӘ¬ІЙУГXRDә°SEM-EDS·Ц±р·ЦОцБЛПаН¬ҢюіцМхәюПВ(ҢюіцОВ¶И30 ҰжҰұЧҒЛЩ200 r/mҰұH2SO4ЕЁ¶И1 mol/LҰұТғ№М±И20:1Ә¬Сх»ҮәБМнәУБү0.5 mol/LӘ¬ҢюіцК±әд6 h)І»Н¬Сх»ҮәБЦЦАаЗү»ҮҢюіцУЛОІүуғуµДОІФьӘ¬·ЦОцҢб№ы·Ц±рәыНә8ҰұНә9ғННә10ЛщКңҰӘ
Нә8ЛщКңОҒҢюіцОІФьµДXRDЖЧҰӘУЙНә8үЙЦҒӘ¬µ±Ңюіц№эіМЦРОөМнәУСх»ҮәБК±Ә¬ҢюіцФьµДXRDЖЧУлФУЛОІүуµДXRDЖЧ»щ±ңПаН¬ҰӘТФMnO2ЧчОҒСх»ҮәБК±Ә¬№иЛбМъСО(Fe3FeSiO4(OH)5)µДМШХч·еГчПФҢµµНӘ¬ЛµГчMnO2ңЯУРЖЖ»µ№иЛбМъСОµДЧчУГӘ¬Н¬К±ЙъіЙБЛРВµД№иЛбГМСО(Mn8Si6O15(OH)10)Ә¬ХвКЗµәЦВҢюіц1.5 hғуУЛҢюіцВКПФЦшПВҢµµДЦчТҒФТтҰӘіэөЛЦ®НвӘ¬ОІФьЦР»№·ұПЦБЛMnO2µДМШХч·еӘ¬¶шMnO2КЗУЛµДёЯР§ОьёҢәБӘ¬ХвТІҢшТ»ІҢЦ¤КµБЛMnO2МнәУ№э¶аИЭТЧµәЦВУЛҢюіцВКПФЦшҢµµНµДПЦПуҰӘМнәУFe3+СОғуµДXRDЖЧУлОөМнәУСх»ҮәБµДXRDЖЧОЮГчПФ±д»ҮӘ¬өУ¶шЦ¤КµБЛFe3+¶Ф№иЛбСООЮЖЖ»µЧчУГӘ¬ХвУлҢюіцКµСйЦРFe3+µДЗү»ҮР§№ыГчПФµНУЪMnO2ПаТ»ЦВҰӘМнәУH2O2µДОІФьXRDЖЧЦР№иЛбМъСО(Fe3FeSiO4(OH)5)µДМШХч·еУРТ»¶ЁµДҢµµНӘ¬ңҰ№ЬЖдЖЖ»µ№иЛбСОµДДЬБ¦ВФІоУЪMnO2Ә¬µ«·өУ¦ғуІ»»бЙъіЙРВµД№иЛбСОүуОпӘ¬өУ¶шҢµµНБЛҢюіцТғЦРУЛФЩөО±»ОьёҢµДёЕВКӘ¬ТтөЛӘ¬ЧоЦХУЛµДҢюіцВКУлH2O2µДМнәУБүіЙХэ±ИӘ¬ХвУлНә7ЦРµДКµСйҢб№ыТ»ЦВҰӘ

Нә8 ҢюіцФьµДXRDЖЧ
Fig. 8 XRD patterns of uranium leaching residue
МнәУІ»Н¬Сх»ҮәБғуҢюіцОІФьµДSEMПсИзНә9ЛщКңҰӘУЙНә9үЙЦҒӘ¬МнәУІ»Н¬Сх»ҮәБғуµДҢюіцОІФьРОГІңЯУРҢПөуµДІоТмҰӘГ»УРМнәУИОғОСх»ҮәБµДҢюіцОІФьТФНЕңЫµДөуүЕБӘРОКҢөжФЪӘ¬ЛµГчҢюіц№эіМЦРҢцүүҢюіцТғ(БтЛб)ОЮ·ЁЖЖ»µЖд±ңЙнµДВцКҮҢб№№Ә¬ХвКЗµәЦВіӘ№жЛбҢюЦРҢюіцК±әді¤ҰұҢюіцР§ВКµНµДЦчТҒФТтҰӘМнәУСх»ҮәБMnO2ғНH2O2ғуӘ¬НЕңЫµДөуүЕБӘГчПФ±діЙҢПРҰіЯөзµДүЕБӘӘ¬ХвТІФЩөОЦ¤ГчБЛMnO2ғНH2O2¶ФВцКҮҢб№№µДЖЖ»µЧчУГӘ¬µ«¶юХЯЦ®әдУЦУРТ»¶ЁµДІо±рҰӘМнәУMnO2µДҢюіцОІФьүЕБӘіЯөзГчПФөуУЪМнәУH2O2µДҢюіцОІФьүЕБӘіЯөзӘ¬ХвКЗТтОҒФЩөОРОіЙ№иЛбГМСО(Mn8Si6O15(OH)10)ғуң§МеіЯөзі¤өуµДФТтӘ¬Н¬К±ТІ·ұПЦБЛЙЩБүОөНкИ«·өУ¦µДMnO2ң§МеҰӘ¶шМнәУH2O2ғуµДОІФьЦРӘ¬іэЙЩБүОөНкИ«±»ЖЖ»µµДөуүЕБӘНвӘ¬УЙУЪ·өУ¦№эіМЦРІ»»бФЩЙъіЙЖдЛыµД№иЛбСОң§МеӘ¬µәЦВүЕБӘµДЖҢңщіЯөзңщҢПРҰӘ¬ТтөЛөЩҢшБЛУЛµДҢвАлӘ¬ХвТІКЗµ±H2O2МнәУБүЧг№»К±Ә¬ЖдЗү»ҮР§№ыУЕУЪMnO2µДЦчТҒФТтҰӘМнәУFe3+ЧчОҒСх»ҮәБК±Ә¬ОІФьµДүЕБӘіЯөзТАңЙҢПөуӘ¬ХвТІФЩөОЛµГчБЛFe3+¶ФВцКҮҢб№№ОЮГчПФµДЖЖ»µЧчУГҰӘіэөЛЦ®НвӘ¬УлОөМнәУСх»ҮәБµДОІФьПа±ИӘ¬үЕБӘ±нГж»№іцПЦБЛМхЧөµДң§МеёІёЗФЪүЕБӘ±нГжӘ¬»б¶ФУЛ»бІъЙъОьёҢЧчУГ(әыНә10 EDS·ЦОцҢб№ы)Ә¬ХвКЗНә7(c)ЦР·өУ¦ғуЖЪУЛҢюіцВКПФЦшҢµµНµДЦчТҒФТтҰӘ

Нә9 ҢюіцОІФьµДSEMПс
Fig. 9 SEM images of uranium leaching residue

Нә10 МнәУІ»Н¬Сх»ҮәБғуҢюіцғуОІФьµДEDS·ЦОцҢб№ы
Fig. 10 EDS results of uranium leaching residue
ҢюіцОІФьµДEDS·ЦОцҢб№ыИзНә10ЛщКңҰӘҢюіцОІФь±нГжµДДЬЖЧФҒЛШЧйіЙ»щ±ңТ»ЦВӘ¬µ«МнәУFe3+ЧчОҒСх»ҮәБµДОІФьЦРУЛғ¬БүГчПФҢПЖдЛыОІФьЖ«ёЯӘ¬ХвТІЦ¤ГчБЛFe3+ЧчОҒСх»ҮәБМнәУБүИз№ы№э¶аӘ¬Ң«»бФЪғуЖЪ¶ФҢюіцТғЦРµДУЛҢшРРОьёҢҰӘЧЫЙП·ЦОцүЙЦҒӘ¬¶ФУЪ3ЦЦСх»ҮәБ¶шСФӘ¬Зү»ҮҢюіцР§№ыЧоғГµДОҒH2O2Ә¬ЖдөООҒMnO2Ә¬ЧоғуОҒFe3+ҰӘИ»¶шӘ¬H2O2ә°MnO2ЖЖ»µВцКҮҢб№№µДОұ№Ы»ъАн»№УРөэУЪЙоИлСРңүҰӘ
3 ҢбВЫ
1) УЛОІүуµДЦчТҒіЙ·ЦОҒёчАа№иЛбСОӘ¬УЛғ¬БүҢцОҒ0.008%ЧуУТӘ¬±ШРлЖЖ»µ№иЛбСОҢб№№ІЕДЬКµПЦУЛµДҢвАлҰӘ
2) ІЙУГіӘ№жБтЛбҢюіц№¤ТХУЛµДҢюіцВКҢПµНӘ¬¶шЗТҢюіцК±әді¤өп6hғуӘ¬УЛµДҢюіцВКҢцОҒ78%ЧуУТҰӘ
3) НЁ№эМнәУСх»ҮәБғуӘ¬УЛµДҢюіцВКә°ҢюіцЛЩ¶ИПФЦшМбёЯӘ¬ФЪ1.5hДЪУЛµДҢюіцВКүЙөп95%Ә¬Сх»ҮәБµДЗү»ҮР§№ыУЕПИЛіРтТАөООҒH2O2ҰұMnO2ғНFe3+ҰӘ
4) H2O2ғНMnO2ФЪҢюіц№эіМЦРµДЗү»ҮЧчУГЦчТҒКЗүүЖЖ»µ№иЛбСОҢб№№»тЙъіЙРВµД№иЛбСОң§Ме¶шКµПЦµДӘ¬Fe3+¶Ф№иЛбСОГ»УРГчПФµДЖЖ»µЧчУГӘ¬ҢцДЬНЁ№эСх»ҮµНәЫУЛЦБёЯәЫУЛАөМбёЯУЛµДҢюіцВКҰӘ
REFERENCES
[1] ҢҒ ОҰ, ёЯОА¶«. µНМәС№Б¦ПВЦР№ъғЛµзІъТµ·ұХ№ә°УЛЧКФө±ӘХП[J]. і¤ҢБчУтЧКФөУл»·ңі, 2011, 20(8): 938-943.
JIANG Wei, GAO Wei-dong. Analysis of China nuclear power industry development and its uranium resource safeguard[J]. Resources and Environment in the Yangtze Basin, 2011, 20(8): 938-943.
[2] ЕӘС§ңь, М·СЗ»Ф, ЛХСЮИг, ЛХС§±у, іВ к». ОТ№ъУЛүуІЙТ±әәКх·ұХ№·ҢПтғНЦШµгИООс[J]. УЛүуТ±, 2013(1): 22-26.
NIU Xue-jun, TAN Ya-hui, SU Yan-ru, SU Xue-bin, CHEN Hao. Development direction and key task of uranium mining and metallurgy techniques in China[J]. Uranium Mining and Metallurgy, 2013(1): 22-26.
[3] Рм АЪ, З®ҢЁЖҢ, МЖЧЁОд. ОТ№ъУЛүу·ПФьКҮОЫИңМШµгә°ЦОАн·Ң·Ё[J]. ЦР№ъүуТµ, 2013, 22(1): 61-64.
XU Lei, QIAN Jian-ping, TANG Zhuan-wu. A study of features and methodology of waste treatment in uranium mines of China[J]. China Mining Magazine, 2013, 22(1): 61-64.
[4] LIU Jin-xiang, XIE Shui-bo, WANG Yong-hua, LIU Ying-jiu, CAI Ping-li, XIONG Fen, WANG Wen-tao. U(ұц) reduction by Shewanella oneidensis mediated by anthraquinone-2-sulfonate[J]. Transactions of Nonferrous Metals Society of China, 2015, 25(12): 4144-4150.
[5] µЛОДңІ, ЦЬКйүы, БхУңЕ, Фш№вГч, ҢғӘғЖ, үµ Ац, ·Ң Бә. ДңРәәңп§тьғПОьёҢәБµДЦЖ±ёә°ЖдОьёҢУЛүуЛб·Ё·ПЛ®ЦРU(ұц)[J]. ЦР№ъУРЙ«ҢрКфС§±Ё, 2015, 25(9): 2604-2611.
DENG Wen-jing, ZHOU Shu-kui, LIU Ying-jiu, ZENG Guang-ming, JIANG Hai-hao, KANG Li, FANG Liang. Preparation of quaternary ammonium salt modified sawdust chelate adsorbent and its U(ұц) adsorption in wastewater from uranium milling plant[J]. The Chinese Journal of Nonferrous Metals, 2015, 25(9): 2604-2611.
[6] KHALED S M, AZAM S. Depositional characteristics of uranium tailings from Saskatchewan, Canada[J]. Environmental Earth Sciences, 2014, 72(11): 4393-4400.
[7] RAO K A, SREENIVAS T, VINJAMUR M, SURI A K. Continuous leaching of uranium from an Indian ore: Residence time scale up and heat effects[J]. Hydrometallurgy, 2014, 146: 119-127.
[8] IBRAHIM M E, LASHEEN T A, HASSIB H B, HELAL A S. Oxidative leaching kinetics of U(ұф) deposit under acidic oxidizing conditions[J]. Journal of Environmental Chemical Engineering, 2013, 1(4): 1194-1198.
[9] COSTINE A, NIKOLOSKI A N, DA COSTA M, CHONG K F, HACKL R. Uranium extraction from a pure natural brannerite mineral by acidic ferric sulphate leaching[J]. Minerals Engineering, 2013, 53: 84-90.
[10] ZAKRZEWSKA K G, HERDZIK K I, COJOCARU C, CHAJDUK E. Experimental design and optimization of leaching process for recovery of valuable chemical elements (U, La, V, Mo, Yb and Th) from low-grade uranium ore[J]. Journal of Hazardous Materials, 2014, 275: 136-145.
[11] SANTOS E A, LADEIRA A C Q. Recovery of uranium from mine waste by leaching with carbonate-based reagents[J]. Environmental Science and Technology, 2011, 45(8): 3591-3597.
[12] OHASHI Y, MURASHITA S, NOMURA M. Extraction of uranium from solid waste containing uranium and fluorine[J]. Minerals Engineering, 2014, 61: 32-39.
[13] TOCINO F, SZENKNECT S, MESBAH A, CLAVIER N, DACHEUX N. Dissolution of uranium mixed oxides: The role of oxygen vacancies vs the redox reactions[J]. Progress in Nuclear Energy, 2014, 72: 101-106.
[14] CHARALAMBOUS F A, RAM R, MCMASTER S, POWNCEBY M I, TARDIO J, BHARGAVA S K. Leaching behaviour of natural and heat-treated brannerite-containing uranium ores in sulphate solutions with iron (ұу)[J]. Minerals Engineering, 2014, 57: 25-35.
[15] LEI Xue-fei, QI Guang-xia, SUN Ying-dong, XU Hui, WANG Yi. Removal of uranium and gross radioactivity from coal bottom ash by CaCl2 roasting followed by HNO3 leaching[J]. Journal of Hazardous Materials, 2014, 276: 346-352.
[16] LADOLA Y S, CHOWDHURY S, ROY S B, PANDIT A B. Application of cavitation in uranium leaching[J]. Desalination and Water Treatment, 2014, 52(1/3): 407-414.
[17] СоУкЙҢ, Уч Зе, ғъ ДП, ¶ҰµВЬ°. ОұІЁәУИИФ¤ө¦Ан¶СҢюУЛүуКҮ[J]. ПҰУРҢрКф, 2016, 40(3): 280- 286.
YANG Yu-shan, YU Qing, HU Nan, DING De-xin. Heap leaching uranium ore pretreated by microwave radiation[J]. Chinese Journal of Rare Metals, 2016, 40(3): 280- 286.
[18] Т¶УВңь, ¶ҰµВЬ°, Ао№гФГ, ·цғӘУӨ, ЛОәь±у, ғъ ДП. І»Н¬БӘң¶·ЦІә·ЦО¬КэУЛүуКҮµДҢюіц№жВЙ[J]. ЦР№ъУРЙ«ҢрКфС§±Ё, 2013, 23(10): 2921-2927.
YE Yong-jun, DING De-xin, LI Guang-yue, FU Hai-ying, SONG Jian-bin, HU Nan. Leaching behavior of uranium ore with different fractal dimensions of particle size distribution[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(10): 2921-2927.
[19] GAO Jia-cheng, WANG Liang-fen, WANG Yong, WU Shu-fang. High-temperature creep properties of uranium dioxide pellet[J]. Transactions of Nonferrous Metals Society of China, 2010, 20(2): 238-242.
[20] YOULTON B J, KINNAIRD J A. GangueЁCreagent interactions during acid leaching of uranium[J]. Minerals Engineering, 2013, 52: 62-73.
[21] ·®Т«Н¤, ВА±ьБб, Рм ҢЬ, ҢҮУА№у, ХЕБХЖә. Л®ИЬТғЦР¶юСх»ҮГМ¶ФУЛµДОьёҢ[J]. »·ңіүЖѧѧ±Ё, 1999, 19(1): 42-46.
FAN Yao-ting,  Bing-ling, XU Jie, JIANG Yong-gui, ZHANG Lin-ping. Adsorption of uranium on manganese dioxide in aqueous solution[J]. Acta Scientiae Circumstantiae, 1999, 19(1): 42-46.
Bing-ling, XU Jie, JIANG Yong-gui, ZHANG Lin-ping. Adsorption of uranium on manganese dioxide in aqueous solution[J]. Acta Scientiae Circumstantiae, 1999, 19(1): 42-46.
[22] WANG Z, LEE S W, CATALANO J G, LEZAMA-PACHECO J S, BARGAR J R, TEBO B M, GIAMMAR D E. Adsorption of uranium (VI) to manganese oxides: X-ray absorption spectroscopy and surface complexation modeling[J]. Environmental Science and Technology, 2012, 47(2): 850-858.
[23] LASHEEN T A, EL-AHMADY M E, HASSIB H B, HELAL A S. Oxidative leaching kinetics of molybdenum-uranium ore in H2SO4 using H2O2 as an oxidizing agent[J]. Frontiers of Chemical Science and Engineering, 2013, 7(1): 95-102.
[24] FAN Fang-li, DING Hua-jie, BAI Jing, WU Xiao-lei, LEI Fuan, TIAN Wei, WANG Yang, QIN Zhi. Sorption of uranium (VI) from aqueous solution onto magnesium silicate hollow spheres[J]. Journal of Radioanalytical and Nuclear Chemistry, 2011, 289(2): 367-374.
[25] WANG Jing-song, BAO Zheng-lei, CHEN Si-guang, YANG Jin-hui. Removal of uranium from aqueous solution by chitosan and ferrous ions[J]. Journal of Engineering for Gas Turbines and Power, 2011, 133(8): 21-23.
Oxidizing leaching of uranium from low-grade uranium tailings
LI Mi1, 2, ZHANG Biao1, ZHANG Xiao-wen1, 2, HUANG Jing1, DING De-xin3, YE Yong-jun3
(1. School of Environment Protection and Safety Engineering, University of South China, Hengyang 421001, China;
2. Key Laboratory of Radioactive Waste Treatment and Disposal, University of South China, Hengyang 421001, China;
3. Key Discipline Laboratory for National Defense for Biotechnology in Uranium Mining and Hydrometallurgy, University of South China, Hengyang 421001, China)
Abstract: In view of low extraction rate of uranium caused by high gangue content and complex occurrence state of phases existing in low-grade uranium tailings, adding suitable oxidants in leaching process to destroy the gangue crystal structure were proposed to enhance the uranium extraction. The effects of traditional acid leaching and intensified leaching with three oxidants (H2O2, MnO2 and Fe3+) on uranium extraction were investigated using single-factor experiments. The results show that uranium extraction ratio is only 78% after 6 h in traditional leaching at temperature, sulfuric acid concentration and liquid-solid ratio of 30 Ұж, 1 mol/L and 20:1, respectively, while the extraction ratio reaches 95% after 1.5 h in intensified leaching under the same leaching conditions. H2O2 and MnO2 can decompose the gangue crystal structure and reduce the particle agglomeration. However, manganous silicate forms when MnO2 is used as oxidants. The ferric iron has no effect on destroying the gangue crystal structure, but it can improve the uranium extraction ratio depending on its oxidation influence.
Key words: uranium tailing; leaching; oxidization; gangue
Foundation item: Project(51404141) supported by the National Natural Science Foundation of China; Project (2015RS4039) supported by the Science and Technology Project of Hunan Province, China; Project (2015M572255) supported by China Postdoctoral Science Foundation; Project(2014KS32) supported by the Science and Technology Project of Hengyang, China
Received date: 2015-09-09; Accepted date: 2016-03-15
Corresponding author: DING De-xin; Tel: +86-734-8282562; E-mail: dingdxzzz@163.com
(±аә Бъ»іЦР)
»щҢрПоДүӘғ№ъәТЧФИ»үЖС§»щҢрЧКЦъПоДү(51404141)Ә»ғюДПКҰөөРВИЛІЕәЖ»®ЧКЦъПоДү(2015RS4039)Ә»ЦР№ъІ©КүғуүЖС§»щҢрЧКЦъПоДү(2015M572255)Ә»ғвСфКРүЖС§әәКх·ұХ№әЖ»®ПоДү(2014KS32)
КХёеИХЖЪӘғ2015-09-09Ә»РЮ¶©ИХЖЪӘғ2016-03-15
НЁРЕЧчХЯӘғ¶ҰµВЬ°Ә¬ҢМКЪӘ¬І©КүӘ»µз»°Әғ0734-8282562Ә»E-mail: dingdxzzz@163.com

