发泡参数对粉末冶金制备泡沫铝显微组织和压缩性能的影响
来源期刊:中国有色金属学报(英文版)2017年第7期
论文作者:T. GERAMIPOUR H. OVEISI
文章页码:1569 - 1579
关键词:泡沫铝;粉末冶金;CaCO3;发泡剂;半开孔显微组织;膨胀;压缩性能
Key words:aluminum foam; powder metallurgy; CaCO3; foaming agent; semi open-cell microstructure; expansion; compressive properties
摘 要:使用廉价CaCO3作为发泡剂,采用粉末压块熔炼工艺制备单胞间具有通道的半开孔泡沫铝。将铝和 CaCO3粉末混合物冷压缩成在空气气氛和一定温度下发泡的致密圆柱形前驱体。研究前驱体压缩压力、发泡剂含量、发泡温度和发泡时间对所得泡沫铝显微组织、线膨胀率、相对密度和压缩性能的影响。结果表明,所得泡沫铝的显微组织分布均匀,晶粒尺寸小于100 μm,具有半开孔结构,相对密度为55.4%~84.4%%。随着压缩压力(127~318)的增大,当发泡剂含量为15%(质量分数)时,泡沫铝的线膨胀率、压缩强度和紧实应变增大。当发泡温度从800 °C升高到1000 °C时,除压缩强度和相对密度外,泡沫铝的其他参数都增大。研究结果表明,最佳发泡温度和发泡时间分别为900 °C和10~25 min。
Abstract: Semi open-cell aluminum foams having channels between individual cells were produced using low cost CaCO3 foaming agent and applying the powder compact melting process. To this end, the aluminum and CaCO3 powder mixtures were cold compacted into dense cylindrical precursors for foaming at specific temperatures under air atmosphere. The effects of several parameters including precursor compaction pressure, foaming agent content as well as temperature and time of the foaming process on the cell microstructure, linear expansion, relative density and compressive properties were investigated. A uniform distribution of cells with sizes less than 100 μm, which form semi open-cell structures with relative densities in the range of 55.4%-84.4%, was obtained. The elevation of compaction pressure between 127-318 MPa and blowing agent up to 15% (mass fraction) led to an increase in the linear expansion, compressive strength and densification strain. By varying the foaming temperature from 800 to 1000 °C, all of the investigated parameters increased except compressive strength and relative density. The results indicated the optimal foaming temperature and time as 900 °C and 10-25 min, respectively.
Trans. Nonferrous Met. Soc. China 27(2017) 1569-1579
T. GERAMIPOUR, H. OVEISI
Department of Materials and Polymer Engineering, Hakim Sabzevari University, Sabzevar 9617976487, Iran
Received 3 June 2016; accepted 28 February 2017
Abstract: Semi open-cell aluminum foams having channels between individual cells were produced using low cost CaCO3 foaming agent and applying the powder compact melting process. To this end, the aluminum and CaCO3 powder mixtures were cold compacted into dense cylindrical precursors for foaming at specific temperatures under air atmosphere. The effects of several parameters including precursor compaction pressure, foaming agent content as well as temperature and time of the foaming process on the cell microstructure, linear expansion, relative density and compressive properties were investigated. A uniform distribution of cells with sizes less than 100 μm, which form semi open-cell structures with relative densities in the range of 55.4%-84.4%, was obtained. The elevation of compaction pressure between 127-318 MPa and blowing agent up to 15% (mass fraction) led to an increase in the linear expansion, compressive strength and densification strain. By varying the foaming temperature from 800 to 1000 °C, all of the investigated parameters increased except compressive strength and relative density. The results indicated the optimal foaming temperature and time as 900 °C and 10-25 min, respectively.
Key words: aluminum foam; powder metallurgy; CaCO3; foaming agent; semi open-cell microstructure; expansion; compressive properties
1 Introduction
Since the first production of metallic foams in 1948 by SOSNICK [1], much attention has been paid to these materials with different compositions, cell microstructures and cell sizes. Their unique properties such as low density, high specific strength, high energy absorption, sound absorption and heat resistance make these materials appropriate for many structural and functional applications such as automotive, aerospace, building industry, filtration, fluid flow control, water purification and acoustic control [2,3].
There are numerous methods to produce metallic foams including sintering of the metal powders or fibers, sintering the metallic hollow spheres, gas injection to the molten metals, casting the molten metals into the polymer foams, powder compact melting, metal vapor deposition on cellular preform, and electro- deposition [2-5]. Among these foaming methods, the powder compact melting process has been widely applied to producing aluminum foams, due to the advantages such as high uniformity of the cells, flexibility in alloy choice without requiring any stabilizer particles and production possibility of near-net shaped parts with complex geometries as well as composite foams [6,7]. This process was first introduced by ALLEN [8] and developed by the Fraunhofer Institute [9,10]. All reports demonstrated that this procedure results in closed-cell foams [6,7,11-18]. Recently, researches have been focused on the control of this process in order to produce foams with improved cell structure, enhanced properties and lower costs. YOU et al [11] investigated the effects of foaming temperature and content of TiH2 foaming agent on the cell structure of aluminum foams produced by powder compact melting process. Their results showed that adjusting these two key factors could lead to the formation of closed-cell foams with uniform cell structure and high porosity. Moreover, SURACE et al [12] investigated the effects of three parameters, i.e., compaction pressure, temperature and SiC content on morphology and compressive properties of aluminum foams produced with TiH2 foaming agent by a powder metallurgy technique. Considering linear expansion, relative density and compressive strength of samples, the optimum set-up parameters in their experiments were recognized as compaction pressure of 430 MPa, foaming temperature of 750 °C and stabilizer content of 3% SiC (mass fraction). KEVORKIJAN et al [13] used CaCO3 and dolomite particles as foaming agents to produce aluminum foams by either powder metallurgy or melt rout. In their experiments, the effects of porosity and density of precursors as well as concentration and morphology of foaming agents on foaming efficiency, relative density and structure of aluminum foams were investigated.
Till now, a large number of studies have been done on the properties of aluminum foams using TiH2 foaming agent and powder metallurgy while CaCO3 foaming agent is cheaper and more controllable. Since there are a few reports on aluminum foams using CaCO3 foaming agent [6,14,15], there is a need to investigate the correlations between processing parameters and final properties of foams produced using CaCO3 as a foaming agent.
In this study, the semi open-cell aluminum foams were produced by powder compact melting process and low cost CaCO3 foaming agent via adjusting the critical processing parameters. Moreover, the effects of these critical parameters including compaction pressure, foaming agent content, foaming temperature and time on linear expansion, relative density, cell microstructure and compressive properties were studied.
2 Experimental
2.1 Materials
Aluminum (Merck, Germany, >99% purity, <15 μm) and CaCO3 (Farzan Powder, Iran, >99.5% purity, <50 μm) powders were used as aluminum source and foaming agent, respectively.
2.2 Samples production
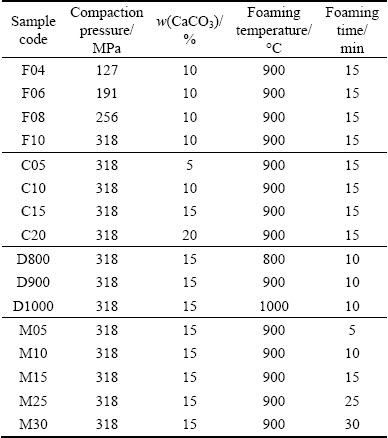
According to “powder compact melting” method, aluminum and CaCO3 powders were mixed together for 20 min with different CaCO3/Al mass ratios (5%, 10%, 15% and 20%) to obtain homogeneous mixtures. The mixed powders were then compacted using uniaxial cold pressing with different pressures (127, 191, 255 and 318 MPa) to form dense cylindrical components called “foamable precursors” with 20 mm in diameter and different heights and densities. The precursors were placed in a cylindrical steel mold (20 mm in inner diameter, 100 mm in height), which was open only at the top. Heat treatment was done in a preheated furnace at different temperatures (800, 900 and 1000 °C) for different time (5, 10, 15, 25 and 30 min) in air atmosphere. The processing parameters of the samples were listed in Table 1. Since the precursors and the mold had the same diameters, expansions occurred only in the height direction. After the foaming process, the samples were removed from the furnace and natural cooling in the air was employed to solidify the foams. The densities of precursors and foam samples were calculated from their mass and geometry. Furthermore, relative density of precursors and foams (ρR), linear expansion (αLE) and porosity of foams (P) were calculated using the following equations [12,19]. It is important to note that all the calculations are in comparison with the bulk aluminum.
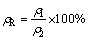 (1)
(1)
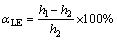 (2)
(2)
where ρ1 and ρ2 are densities of precursor or foam and bulk aluminum, respectively; h1 and h2 are heights of foam and precursor, respectively.
Table 1 Processing parameters of produced samples
2.3 Characterization
In order to determine the decomposition temperature range of the foaming agent (CaCO3), differential thermal analysis (DTA) was performed. The pattern was recorded on BAHR Thermo Analyse 703 machine from 25 to 1000 °C with a heating rate of 10 °C/min in argon atmosphere. The CaCO3 (35 mg) and alumina (35 mg) powders were used as foaming agent and the reference material, respectively. In order to compare the compressive data, samples were cut into cylindrical parts with 20 mm in diameter and height to diameter ratios in the range of 1.5-2.0. The minimum dimension of the specimen was at least seven times of the cell size to avoid size effects [3]. The compression tests were performed at room temperature using a Zwick/ Roell Z250 testing machine at the constant ram speed of 6 mm/min. Furthermore, samples were sectioned using a wire-cutting machine and the cell microstructure was observed with light microscope (LM, OLYMPUS GX51) and field emission scanning electron microscope (FESEM, TESCAN Mira 3-XMU) using an accelerating voltage of 15.0 kV.
3 Results and discussion
3.1 Thermal properties of foaming agent
In order to determine the thermal properties of foaming agent, the DTA analysis was carried out. The DTA pattern of CaCO3 powder shown in Fig. 1 indicates two endothermic peaks at 30-250 °C and 670-900 °C, which are related to the moisture loss and CaCO3 decomposition reactions, respectively. The thermal decomposition and CO2 gas release started at approximately 670 °C which was completed at about 900 °C. The maximum decomposition rate (Rmax) of the CaCO3 foaming agent was at 850 °C. Hence, three different temperatures, 800 °C (between decomposition and Rmax temperature), 900 °C (temperature of complete decomposition) and 1000 °C (above the decomposition temperature) were selected for the foaming process. Furthermore, at temperatures above 400 °C, the baseline declined due to the increase of heat capacity difference between CaCO3 foaming agent and Al2O3 reference.
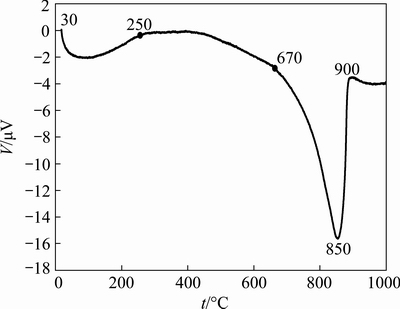
Fig. 1 DTA pattern of CaCO3 foaming agent

Fig. 2 Images of produced samples with different compaction pressures (a, b), foaming agent contents (c, d), foaming temperatures (e, f) and foaming time (g, h) before and after heat treatment
3.2 Determination of porosity, linear expansion coefficient and relative density
Figure 2 shows the images of produced samples using different compaction pressures (Figs. 2(a) and (b)), CaCO3 foaming agent contents (Figs. 2(c) and (d)), foaming temperatures (Figs. 2(e) and (f)) and foaming time (Figs. 2(g) and (h)). The large sectional cracks which were observed in some foam samples were related to the formation of shear bands due to higher stress in the upper and lower zones in the precursors during compaction. Therefore, when the punch and die moved away from each other, elastic recovery of these two areas resulted in the formation of sectional shear bands inside the precursors [19]. These cracks became obvious during foaming and expanding of precursors.
Table 2 Porosity, linear expansion and relative density of foam samples
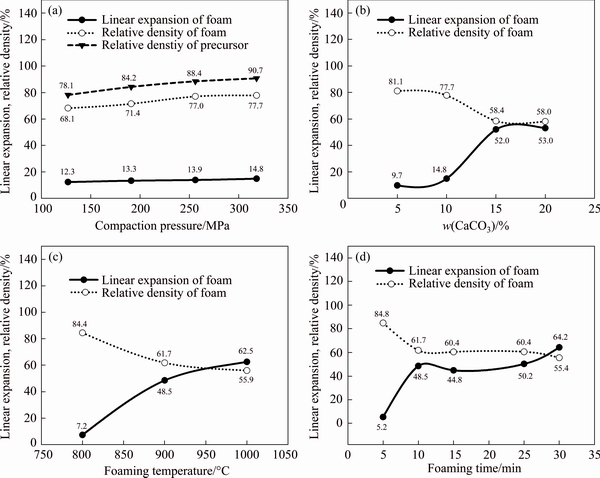
Fig. 3 Effects of compaction pressure (a), CaCO3 foaming agent content (b), foaming temperature (c) and foaming time (d) on foaming behavior
The porosity, linear expansion and relative density of all samples are listed in Table 2. According to Table 2, samples C15, C20 and M30 showed the highest porosity (41.9%, 41.9% and 44.5%, respectively), linear expansion (52.0%, 53.0% and 55.4%, respectively) and the lowest relative density (58.4%, 58.0% and 55.4%, respectively).
The linear expansion and relative density of the precursors and foam samples with different processing parameters are shown in Fig. 3. It can be seen from Fig. 3(a) that increasing the compaction pressures from 127 up to 318 MPa resulted in the denser precursors and enhancement of foam linear expansion and relative density from 12.3% to 14.8% and 68.1% to 77.7%, respectively. Therefore, by increasing the compaction pressure, the released CO2 gas in the foaming step was increasingly entrapped in the precursors and more linear expansions (up to 14.8%) were obtained. In other words, higher precursor density with compaction pressure resulted in more easily foaming due to lower remaining interconnected pores in the precursors [12,20,21].
As shown in Fig. 3(b), increasing CaCO3 content from 5% to 10% and 15% to 20% (mass fraction) had only a small effect on the linear expansion and relative density of foams while increasing CaCO3 content from 10% to 15%, dramatically increased the linear expansion (14.8% to 52.0%) and decreased the relative density (77.7% to 58.4%).
Increasing the foaming temperature from 800 to 1000 °C enhanced the linear expansion from 7.2% to 62.5%, while decreased the relative density from 84.4% to 55.9%. According to the DTA curve of CaCO3 powder (Fig. 1), the maximum decomposition rate (Rmax) occurred at 850 °C. Clearly, the low linear expansion and high relative density of samples at 800 °C (Fig. 3(c)) were due to the incomplete decomposition of CaCO3. Increasing the foaming temperature resulted in faster CaCO3 decomposition, higher linear expansion and lower relative density. On the other hand, because of the low relative density of the precursors (~90.7%), oxygen could diffuse during heat treatment. Enhanced oxidation of aluminum matrix with temperature improved the viscosity and stability of foams, which led to higher linear expansions [22]. Particularly in the case of CaCO3 foaming agent, thicker oxide layers on the pore surfaces compared to TiH2 agent helped the stabilization of the foam and inhibited the collapse of the microstructure [23].
(Fig. 3(c)) were due to the incomplete decomposition of CaCO3. Increasing the foaming temperature resulted in faster CaCO3 decomposition, higher linear expansion and lower relative density. On the other hand, because of the low relative density of the precursors (~90.7%), oxygen could diffuse during heat treatment. Enhanced oxidation of aluminum matrix with temperature improved the viscosity and stability of foams, which led to higher linear expansions [22]. Particularly in the case of CaCO3 foaming agent, thicker oxide layers on the pore surfaces compared to TiH2 agent helped the stabilization of the foam and inhibited the collapse of the microstructure [23].
When precursors were heated at 900 °C for 5 min, the linear expansion was very low (5.2%), indicating the insufficient time for decomposition of CaCO3. Therefore, the relative density of the 5 min-heated foam was as high as 84.8%. Increasing the foaming time from 5 to 10 min resulted in a rapid increase of the linear expansion (5.2% to 48.5%) and consequently a rapid decrease of the relative density (84.8% to 61.7%). The foaming time between 10 and 25 min resulted in almost a constant linear expansion and relative density in the range of 48.5%-50.2% and 61.7%-60.4%, respectively. This was due to the complete decomposition of CaCO3. Increasing the foaming time to 30 min resulted in the foams with higher linear expansion (64.2%) and lower relative density (55.4%). Collapse of foam structure and increase in density can be observed in Al-Si foam with TiH2 foaming agent after 350-400 s foaming time [24], while in this research using CaCO3 foaming agent, increasing of relative density is observed even at foaming time of 30 min. This may be due to different stabilizing mechanisms, including existing oxide layer on aluminum particles, further oxidation of aluminum during foaming and CaCO3 decomposition product (i.e., CaO); all of these solids, especially CaO, helped the foam stability via increasing the viscosity [22,25-28].
3.3 Cell microstructure
In order to investigate the cell microstructure of the samples, LM and FESEM were used. Figure 4 shows the optical micrographs of F04, F06, F08, F10, C10, C15 and C20 samples with different compaction pressures and foaming agent contents. The dark areas indicate the porosities, which are material-free regions that cannot reflect the light. Since cell walls were very thin, some of them were damaged during the wire-cutting process. The circles show some of these damaged regions. As it can be seen in Fig. 4, increasing compaction pressure resulted in more uniformity of cell size and distribution (Figs. 4(a)-(c)). Figures 4(d)-(f) respectively show the images of C10, C15 and C20 samples. The cell microstructures (size and distribution) of the C10 and C15 samples were relatively uniform. The agglomeration of CaCO3 particles as well as massive gas released during foaming in the C20 sample resulted in the formation of unusual large cells and non-uniform cell microstructure.
Figure 5 shows cross sectional FESEM images of the D1000 sample with porosity of 44.0% and relative density of 55.9%. As it can be seen in Fig. 5, the aluminum particles were sintered during foaming treatment and formed the cell walls. The uniform distribution of the cells, which connected via some channels, was observed. In fact, the size of Al and CaCO3 powder caused the individual pores to join and form some channels between the cells, which are indicated by arrows (Figs. 5(c) and (d)). However, since each cell did not have at least two open ends, the microstructure can be considered as semi open-cell. The cell sizes were smaller than 100 μm with irregular shapes, which was due to very fine aluminum particles [29]. In Fig. 5(d), small pores between primary aluminum powders indicated by dotted-line circles were due to powder compact melting process. These pores related to the presence of the oxide layer on the surface of very fine aluminum particles, which deteriorated the sufficient diffusion and complete bonding of the particles. The full-line circle shows that partial melting has occurred during the foaming process (Fig. 5(d)). The formation of big pores in millimeter dimensions (Fig. 5(a)) is due to the agglomerated CaCO3 particles and consequently a massive gas release during the foaming process. The elongated shape of these pores is due to flattening of the agglomerated CaCO3 particles during compacting of powder mixtures [30].

Fig. 4 Light micrographs of F06 (a), F08 (b), F10 (c), C10 (d), C15 (e) and C20 (f) samples
Different steps of the foam formation are shown schematically in Fig. 6. Figure 6(a) shows a mixture of aluminum (black particles) and CaCO3 (gray particles) powders. There was a thin oxide layer on the surface of most aluminum particles and some parts of these oxide layers were broken down during cold pressing. The aluminum particles were cold-welded together and a dense material, which is known as the precursor, was obtained (Fig. 6(b)). As indicated in Fig. 6(c), heating the precursor up to a specific temperature resulted in releasing CO2 gas and partially melting of aluminum particles. As it can be seen in this figure, the particle size ratio of aluminum to CaCO3 allowed the possibility of connecting individual large pores and formation of some channels, which led to semi open-cell microstructure. During heating the precursor, aluminum particles were partially melted and sintered throughout the cold-welded sites, but aluminum oxide layers inhibited connecting the melted aluminum particles. Hence, at foaming temperatures as high as 800-1000 °C, aluminum particles were sintered together and remained separate.

Fig. 5 FESEM images of D1000 sample with different magnifications

Fig. 6 Schematic illustration of foam formation steps (The white arrows show small pores due to powder metallurgy while black arrows and circles show pores due to CaCO3 decomposition and channels between individual cells, respectively)
3.4 Compressive properties
Figure 7 shows the compressive stress-strain curves of the samples produced with different processing parameters. Researchers usually consider three regions of elastic, plateau and bulk behavior for compressive curves of metallic foams [3,31-33]. However, OCHSNER and LAMPRECHT [34] have shown that there is a region of elastic to plastic transition in these curves. Hence, four consecutive regions for compressive curves in Fig. 7 can be considered. These regions which have been marked on the curve of the F10 sample include 1) elastic deformation (cell walls underwent elastic deformation and the stress increased linearly with the increase of strain), 2) transition zone from elastic to plastic deformation (at the end of this region, the foam began plastic deformation and the stress level was considered as compressive strength (σc), 3) plateau region (a linear region, in which cell walls plastically deformed, buckled and collapsed. The strain at the end of this region is named densification strain (εD)) and 4) close contact of cell walls and collapse (in the final region, all cell walls were in contact with each other and no porosity remained in the structure; and in this region, the sample behaved as a bulk material.
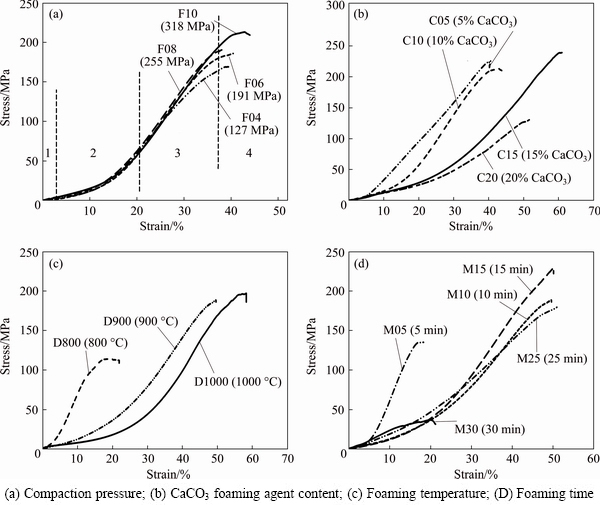
Fig. 7 Compressive stress-strain curves of produced samples with different processing parameters (Four consecutive regions are marked on curve of F10 sample in Fig. (a))
In the plateau region of compressive curves in Fig. 7, the stress linearly increased with the increase of strain. This behavior, which is more typical for open-cell foams, was due to the strain hardening of aluminum and lack of specific cell walls to stabilize the compression [35]. Furthermore, in all compressive curves in Fig. 7, high compressive stress, intensive strain hardening and short collapse regions (regions 3 and 4) were observed, which represented the brittle behavior of aluminum foams in compression. Two main factors can be considered for this observation: first, the small pores between primary aluminum particles due to powder metallurgy (PM pores) and second, different solid particles such as CaO (product of CaCO3 decomposition), un-decomposed CaCO3 and Al2O3 (broken oxide layer on aluminum particles) in the structure. These PM pores and brittle particles with low plasticity acted as likely sites for crack initiation and brittle behavior of aluminum foams [31,36].
As shown in Fig. 7(a), increasing of compaction pressure resulted in a longer plateau region. Figure 7(b) demonstrates that as CaCO3 content increased, compressive stress significantly decreased and samples with CaCO3 content of 15% showed the highest compressive stress and strain (235 MPa and 60%, respectively). In Fig. 7(c), it is obvious that increasing foaming temperature from 800 to 1000 °C enhanced the compressive stress (110 to 190 MPa) and strain (18% to 55%) rapidly. This was due to a higher diffusion, more interfaces between aluminum particles and higher strength of the cell walls [29]. As it can be seen in Fig. 7(d), the sample which was heated for 5 min collapsed at low compressive stress (≈135 MPa) and strain (≈17%). When foaming time was increased to 25 min, compressive stress (175 MPa) and strain (50%) significantly increased. This enhancement was likely due to the complete decomposition of CaCO3 during the foaming process. Increasing the foaming time up to 30 min resulted in a dramatic decrease of compressive stress (≈35 MPa) and strain (≈20%). Therefore, the optimum foaming time was 10-25 min, considering the compressive stress and strain.
Figure 8 shows the plotted data which were extracted from Fig. 7. The increase of compaction pressure from 127 to 318 MPa in Fig. 8(a) resulted in higher elastic modulus (0.9 to 1.5 MPa), compressive strength (19.4 to 32.7 MPa) and densification strain (30% to 40%). This enhancement in compressive properties was due to uniformity of cell microstructure (Figs. 4(a)-(d)) which was improved with compaction pressure. In fact, in foams with uniform cell microstructure, deformation can take place throughout the whole specimen rather than the larger cells. Therefore, higher compressive strength can be obtained [37]. The compressive strength and densification strain were enhanced from 9.4 to 44.1 MPa and 32% to 59% by increasing the amount of CaCO3 from 5% to 15%, respectively (Fig. 8(b)). Further addition of CaCO3 to 20% resulted in the reduced compressive strength (32.5 MPa) and densification strain (40%). This may be due to the agglomeration of CaCO3 particles as well as the massive CO2 gas released during foaming which resulted in the formation of unusually large cells, non- uniform cell microstructure (Fig. 4(g)) and consequently in lower compressive properties [38]. Since the maximum decomposition rate of CaCO3 occurred at 850 °C (Fig. 1), heating the precursors at 800 °C resulted in insufficient decomposition of CaCO3 and consequently undesired compressive properties (Fig. 8(c)). Increasing the foaming temperature from 800 to 900 °C resulted in rapid increases of compressive strength from 13.6 to 40.4 MPa and densification strain from 12% to 47%. At temperature as high as 1000 °C, compressive strength decreased to 35.6 MPa and densification strain gradually increased to 48%. In Fig. 8(d), at a foaming time of 10-25 min, maximum compressive strength (40.5-45.0 MPa) and densification strain (45%-47%) were obtained. The compressive properties were low for time periods less than 10 min and more than 25 min due to insufficient decomposition of CaCO3 and collapsing of cells, respectively.
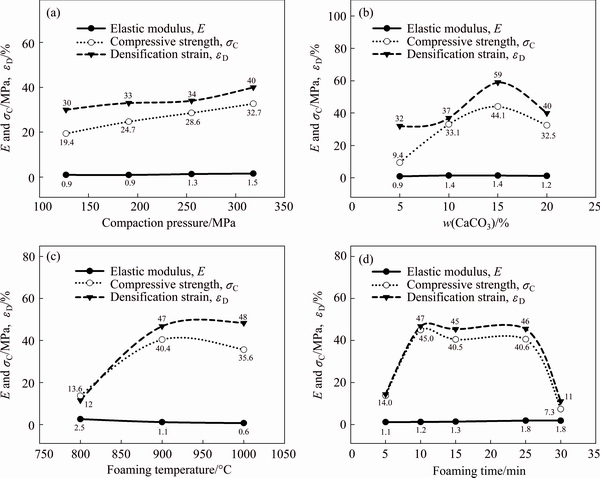
Fig. 8 Effects of compaction pressure (a), CaCO3 foaming agent content (b), foaming temperature (c) and foaming time (d) on compressive properties
4 Conclusions
1) A facile method based on the powder metallurgy was used to produce aluminum foam. Adjusting the parameters of the process such as particle size and metal/foaming agent ratio resulted in joining individual pores and forming some channels between cells that led to semi open-cell microstructure. Step-by-step optimization of compaction pressure, foaming agent content, foaming temperature and time was performed.
2) Increasing the compaction pressure from 127 to 318 MPa improved the cell microstructure and enhanced the relative density (68.1%-77.8%) and compressive strength (19.4-32.7 MPa).
3) CaCO3 content of 15% was the optimum content for foaming agent in which the lowest relative density (58.4%), uniform cell microstructure and desired compressive strength (44.1 MPa) were obtained.
4) Increasing the foaming temperature from 800 to 1000 °C lowered the relative density (84.4%-55.9%) and improved the compressive strength (13.6-35.6 MPa).
5) The compressive strength was low in time periods less than 10 min and more than 25 min due to insufficient CaCO3 decomposition and collapsing of cells. For the foaming time of 10-25 min, the maximum compressive strength (40.5-45.0 MPa) and relative density (60.4%-61.7%) were obtained.
References
[1] SOSNICK B. Process for making foamlike mass of metal: US patent, 2434775 [P]. 1948-01-20.
[2] BANHART J. Manufacture, characterization and application of cellular metals and metal foams [J]. Progress in Materials Science, 2001, 46: 559-632.
[3] ASHBY M F, EVANS A G, FLECK N A, GIBSON L J, HUTCHINSON J W, WADLEY H N G. Metal foams: A design guide [M]. Oxford: Butterworth-Heinemann, 2000.
[4] DAVIES G J, ZHEN S. Metallic foams: Their production, properties and applications [J]. Journal of Materials Science, 1983, 18: 1899-1911.
[5] BANHART J, BAUMEISTER J. Production methods for metallic foams [J]. Materials Research Society, 1998, 521: 121-132.
[6] YU C J, EIFERT H H, BANHART J, BAUMEISTER J. Metal foaming by a powder metallurgy method: Production, properties and applications [J]. Material Research Innovations, 1998, 2: 181-188.
[7] BAUMGARTNER F, DUARTE I, BANHART J. Industrialization of powder compact foaming process [J]. Advanced Engineering Materials, 2000, 2: 168-174.
[8] ALLEN B C. Method of making foamed metal: US patent, 3087807 [P]. 1963-04-30.
[9] BAUMEISTER J. Porous metal body production-involves compaction at low temperature followed by heating to near melting point of metal: German patent, 4018360 [P]. 1991-05-29.
[10] BAUMEISTER J, SCHRADER H. Methods for manufacturing foamable metal bodies: German patent, 4101630 [P]. 1992-04-16.
[11] YOU X H, WANG F, WANG L C. The structure control of aluminum foams produced by powder compact foaming process [J]. Acta Metallurgica Sinica, 2004, 17: 279-282.
[12] SURACE R, de FILIPPIS L A C, LUDOVICOA A D, BOGHETICHC G. Experimental analysis of the effect of control factors on aluminium foam produced by powder metallurgy [J]. Proceedings of the Estonian Academy of Sciences Engineering, 2007, 13: 156-167.
[13] KEVORKIJAN V, SKAPIN S D, PAULIN I, SUSTARSIC B, JENKO M, LAZETA M. Influence of the foaming precursor’s compaction and density on the foaming efficiency, microstructure development and mechanical properties of aluminum foams [J]. Materials and Technology, 2011, 45: 95-103.
[14] DUARTE I, BANHART J. A study of aluminum foam formation kinetics and microstructure [J]. Acta Materialia, 2000, 48: 2349-2362.
[15] BABCSAN N, BANHART J, LEITLMEIER D. Metal foams―Manufacture and physics of foaming [C]//JERZ J. Advanced Metallic Materials 2003: Proceedings of the International Conference. Bratislava: Institute of Materials & Machine Mechanics Slovak Academy of Science, 2003: 5-15.
[16] PAULIN I. Synthesis and characterization of Al foams produced by powder metallurgy route using dolomite and titanium hydride as a foaming agents [J]. Materials and Technology, 2014, 48: 943-947.
[17] FENG Y, ZHENG H W, ZHU Z G, ZU F Q. The microstructure and electrical conductivity of aluminum alloy foams [J]. Materials Chemistry and Physics, 2012, 78: 196-201.
[18] YOUN S W, KANG C G. Fabrication of foamable precursors by powder compression and induction heating process [J]. Metallurgical and Materials Transaction B, 2004, 35: 769-776.
[19] GILANI H, JAFARI S, GHOLAMI R, HABIBOLAHZADEH A, MIRSHAHI M. Effect of processing parameters and glycerin addition on the properties of Al foams [J]. Metals and Materials International, 2012, 18: 327-333.
[20] KENNEDY A R. Effect of compaction density on foamability of Al-TiH2 powder compacts [J]. Powder Metallurgy, 2002, 45: 75-79.
[21] PAULIN I, SUSTARSIC B, KEVORKIJAN V, SKAPIN S D, JENKO M. Synthesis of aluminium foams by the powder-metallurgy process: Compacting of precursors [J]. Materials and Technology, 2011, 45: 13-19.
[22] ASAVAVISITHCHAI S, KENNEDY A R. The effect of compaction method on the expansion and stability of aluminum foams [J]. Advanced Engineering Materials, 2006, 8: 810-815.
[23] PAULIN I. Stability of close-cell Al foams depending on the usage of different foaming agents [J]. Materials and Technology, 2015, 49: 983-988.
[24] SEIFI S M, TABATABAEI Y, MIRBAGHERI S M H, TAFTEH R. Effects of addition of SiC and TiB2 particles on structural and mechanical properties of PM Al-Si foams [C]//Proceedings of Materials Science & Technology Conference and Exhibition 2011 (MS&T 11). Warrendale, PA: Association for Iron & Steel Technology, 2011: 859-866.
[25] GERGELY V, CURRAN D C, CLYNE T W. The FOAMCARP process: Foaming of aluminium MMCs by the chalk-aluminium reaction in precursors [J]. Composites Science and Technology, 2003, 63: 2301-2310.
[26] BRYANT J D, CROWLEY M, WANG W, WILHELMY D, KALLIVAYALI J. Development of Alcoa aluminum foam products [C]//Proceedings of the Fifth International Conference on Porous Metals and Metallic Foams. Lancaster, PA: Destech Publisher, 2008: 19-22.
[27] BANHART J. Metal foams: Production and stability [J]. Advanced Engineering Materials, 2006, 8: 781-794.
[28] HAIBEL A, RACK A, BANHART J. Why are metal foams stable? [J]. Applied Physics Letters, 2006, 89: 154102.
[29] CHINO Y, MABUCHI M, NAKANISHI H, IWASAKI H, YAMMOTO A, TSUBAKINO H. Effect of metal powder size on the gas expansion behavior of 7075 Al alloy in a semisolid state [J]. Materials Science and Engineering A, 2004, 382: 35-40.
[30] HELFEN L, BAUMBACH T, STANZICK H, BANHART J, ELMOUTAOUAKKIL A, CLOETENS P. Viewing the early stage of metal foam formation by computed tomography using synchrotron radiation [J]. Advanced Engineering Materials, 2002, 4: 808-813.
[31] MIRZAEE M, CHANGIZI R, ALINEJAD B. Comparison of mechanical and electrical properties of foams fabricated by the methods of sinter followed cold press and hot press [J]. ARPN Journal of Engineering and Applied Sciences, 2012, 7: 1533-1538.
[32] KOZA E, LEONOWICZ M, WOJCIECHOWSKI S, SIMANCIK F. Compressive strength of aluminum foams [J]. Materials Letters, 2003, 58: 132-135.
[33] SHARMA V, GHOSE J, KUMAR S. Compressive and acoustic behavioural analysis of Al-MMC foam for industrial applications [J]. Journal of the Institution of Engineers C (India), 2012, 93: 33-40.
[34] OCHSNER A, LAMPRECHT K. On the uniaxial compression behavior of regular shaped cellular metals [J]. Mechanics Research Communications, 2003, 30: 573-579.
[35] ASAVAVISITHCHAI S, PREUKSARATTANAWUT T, NISARATANAPOM E. Microstructure and compressive properties of open-cell silver foams with different pore architectures [J]. Procedia Materials Science, 2014, 4: 51-55.
[36] BYAKOVA A, GNYLOSKURENKO S, NAKAMURA T. The role of foaming agent and processing route in the mechanical performance of fabricated aluminum foams [J]. Metals, 2012, 2: 95-112.
[37] WANG D Q. Relation of cell uniformity and mechanical property of a close cell aluminum foam [J]. Advanced Engineering Materials, 2013, 3: 175-179.
[38] SAADATFAR M, MUKHERJEE M, MADADI M, SCHRODER-TURK G E, GARCIA-MORENO F, SCHALLER F M, HUTZLER S, SHEPPARD A P, BANHART J, RAMAMURTY U. Structure and deformation correlation of closed cell aluminium foam subject to uniaxial compression [J]. Acta Materialia, 2012, 60: 3604-3615.
T. GERAMIPOUR, H. OVEISI
Department of Materials and Polymer Engineering, Hakim Sabzevari University, Sabzevar 9617976487, Iran
摘 要:使用廉价CaCO3作为发泡剂,采用粉末压块熔炼工艺制备单胞间具有通道的半开孔泡沫铝。将铝和CaCO3粉末混合物冷压缩成在空气气氛和一定温度下发泡的致密圆柱形前驱体。研究前驱体压缩压力、发泡剂含量、发泡温度和发泡时间对所得泡沫铝显微组织、线膨胀率、相对密度和压缩性能的影响。结果表明,所得泡沫铝的显微组织分布均匀,晶粒尺寸小于100 μm,具有半开孔结构,相对密度为55.4%~84.4%%。随着压缩压力(127~318)的增大,当发泡剂含量为15%(质量分数)时,泡沫铝的线膨胀率、压缩强度和紧实应变增大。当发泡温度从800 °C升高到1000 °C时,除压缩强度和相对密度外,泡沫铝的其他参数都增大。研究结果表明,最佳发泡温度和发泡时间分别为900 °C和10~25 min。
关键词:泡沫铝;粉末冶金;CaCO3;发泡剂;半开孔显微组织;膨胀;压缩性能
(Edited by Wei-ping CHEN)
Corresponding author: H. OVEISI; Tel: +98-51-44012778; E-mail: hamid.oveisi@hsu.ac.ir
DOI: 10.1016/S1003-6326(17)60178-X

