金属Ti热学性质第一原理研究
聂耀庄1, 2,谢佑卿2,彭红建2,李小波2
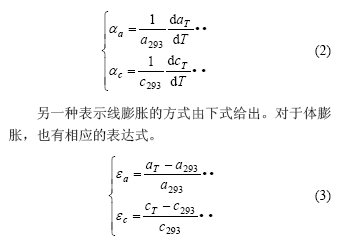
(1. 中南大学 物理科学与技术学院,湖南 长沙,410083;
2. 中南大学 材料科学与工程学院,湖南 长沙,410083)
摘 要:应用基于密度泛函与密度泛函微扰理论的平面波赝势方法计算一组不同晶格常数下六角密堆积(hcp)结构金属Ti的声子谱及相应的静态总能,由此得到不同晶格常数下的自由能,由准谐近似及自由能极小判据得到自由能与温度的关系,进而计算热膨胀系数、定容摩尔热容及定压摩尔热容与温度的关系,对热膨胀系数及定容摩尔热容的第一原理计算值与德拜理论计算值进行比较。结果表明:295 K下声子谱理论值与实验值除在[001]方向上的光学纵模有少量偏差外,其余部分符合得很好;hcp结构金属Ti有一定程度的各向异性热膨胀,沿c轴方向与a轴方向的热膨胀系数比值为1.5左右;热膨胀系数、定容摩尔热容及定压摩尔热容第一原理计算值在较宽的温度范围内与已有的实验数据相符;热膨胀系数的德拜理论值仅在室温以下温区与实验结果相符;定容摩尔热容的第一原理计算值与德拜理论值在中温区有少量偏差,在低温及高温区非常接近。
关键词:第一原理计算;热学性质;声子
中图分类号:O621.24 文献标识码:A 文章编号:1672-7207(2007)06-1072-06
First-principles study of thermal properties of metal Ti
NIE Yao-zhuang1, 2, XIE You-qing2, PENG Hong-jian2, LI Xiao-bo2
(1. School of Physics Science and Technology, Central South University, Changsha 410083, China;
2. School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: A first-principles study of thermal properties of hexagonal close-packed (hcp) metal Ti using the pseudopotential plane-wave method in the framework of the density-functional theory and density-functional perturbation theory was reported. The phonon spectrum and static total energy were calculated at a set of different lattice parameters and free energies at different temperatures were obtained in the quasiharmonic approximation according to the criterion of free energy minimum. The curves of the coefficients of thermal expansion (CTE) and molar heat capacities at constant volume (pressure) to temperature were presented. The coefficients of thermal expansion and molar heat capacities obtained from first-principles approach and Debye theory were compared. The results show that the phonon spectrum at 295 K is in good agreement with available experimental data except the longitudinal optic mode in the [001]; the ratio of CTE along c axis and a axis is about 1.5; CTE and molar heat capacities obtained from first-principles approach are in good agreement with available experimental data in a wide range of temperature; CTE obtained from Debye theory only agrees with experiment below ambient temperature; molar heat capacities at constant volume calculated from first-principles approach and Debye theory have small discrepancy in medial temperature zone.
Key words: first-principles calculations; thermal properties; phonons
基于密度泛函理论的第一原理方法已经成功地应用于不同体系的基态电子结构及物理性能的计 算[1-2]。而对材料晶格动力学的第一原理计算,直到最近10余年才有较大进展。固体的热学性质,如热容和热膨胀,与它们的晶格动力学行为有关。一个简化的计算方案是对晶格动力学行为采用德拜-格林奈森模型[3-4],基于这个方案,人们计算了一些金属[5-6]和化合物[7]的热学性质。在这些工作中,静态总能由第一原理方法算出,晶格振动自由能则由德拜模型算出,德拜温度、格利奈森参数与体积的关系由适当的简化模型给出,由此得出体系的热学性质。这类方法不需要计算体系的声子谱,对热学性质的计算并不完全是基于第一原理的。而冻声方法与分子动力学方法则由于计算量巨大而使它们的应用受到限制。20世纪末发展的密度泛函微扰理论[8-9]使晶格动力学计算有了突破性进展。与其他方法相比,密度泛函微扰理论能够以较小的计算量精确计算布里渊区任一点的声子频率,这使它得到广泛应用。现在已有越来越多的基于密度泛函微扰理论的晶格动力学计算。然而,当计算热学性质如热膨胀时,需在不同晶格参数下进行晶格动力学计算,计算量依然很大。目前,只有少数文献[10-13]报道了材料的热膨胀计算,而且主要限于立方晶系。这是因为,对于非立方晶系,晶格常数有几个分量,这时计算量非常大。对非立方系材料的热膨胀计算仅见于文献[14]。在此,本文作者应用密度泛函微扰理论计算hcp结构金属Ti的定容摩尔热容、定压摩尔热容及各向异性热膨胀等物理量的温度变化曲线。
1 计算方法
采用ABINIT[15]软件包计算Ti的声子谱。ABINIT是基于密度泛函理论及密度泛函微扰理论的平面波赝势软件包。使用Troullier-Martins模守恒赝势[16],交换关联势采用局域密度近似(LDA)。截断能量为980 eV,K点网格为10×10×6,声子频率的计算在5×5×3的q点网格上进行。这些参数的选择使得总能量收敛到1 meV的水平。
对于给定的晶格常数,计算出声子谱,进而可由式(1)得到不同温度时的自由能。

由准谐近似,原子间相互作用的非谐效应体现在不同晶格常数下晶体声子谱不同。计算热膨胀系数需求出不同晶格常数下的声子谱,得出相应的自由能与温度关系,在特定温度下,求自由能的极小值对应的晶格常数,即为该温度下的平衡晶格常数。对hcp结构Ti,令晶格常数a与c与0 K时的a0与c0比值a/a0与c/c0分别由0.99变化至1.02,变化步长为0.5%,共取49组晶格常数值,分别计算出相应的自由能。再由二维样条插值,得到任意晶格常数的自由能与温度关系。通过求极小,得出任意温度下的平衡晶格常数。由式(2)既可求出线热膨胀系数。
由声子谱可以计算振动自由能,进而可以由式(4)求出

根据热膨胀计算结果,选取不同温度下的平衡晶格常数,计算相应的弹性常数,得出体弹性模量B,再由式(5)即可得到 
为与第一原理方法进行比较,下面给出根据德拜-格林奈森模型求热膨胀的方法。只要求出弹性常数,就可以计算其它任何弹性物理量,如体弹性模量、剪切模量、杨氏模量及柏松比等,并且可以由克里斯托弗(Christoffel)方程[17]给出沿任意方向的弹性波速。对大量不同方向的波速求平均值,得到平均声速,进而根据德拜模型,由下式 

2 计算结果
为测试所使用的赝势,计算了0 K下的平衡晶格常数及体弹性模量,计算结果见表1。从表1可看出,c的理论值与实验值相对误差小于0.2%,a的相对误差为1.7%左右,体弹性模量相对误差小于4%。计算结果表明,该赝势符合要求。
表 1 金属Ti的平衡晶格常数及体弹性模量
Table 1 Lattice constants and bulk modulus of Ti

图1所示为在一些高对称方向上室温声子谱理论与实验值[19]的对比结果,可以看到理论结果与实验结果较符合,特别是声学支,理论结果与实验结果符合得很好。在[001]方向上光学支纵模(高频部分)的理论计算与实验偏差相对大一些。考虑到温度不太高时,低频振动模对热物理性质的作用更大,这一部分(光学支)理论与实验的偏差对后面的计算影响应该不大。

1―理论曲线;2―实验值
图 1 hcp Ti声子色散曲线
Fig.1 Dispersion curves of hcp Ti phonon
图2所示为体膨胀的理论值与实验值。可见,在较宽的温度范围内(0~600 K),体膨胀(ε)的理论值与实验值基本相同。在高于1 000 K时,随着温度增加,理论值与实验值的偏差显著增加。与体膨胀相比,体膨胀系数理论曲线与实验值的偏差稍大。虽然如此,在相当宽的温度范围内理论计算值也与实验值基本相
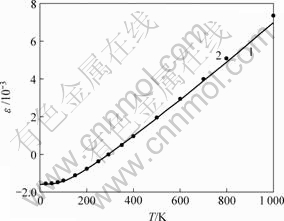
1―理论曲线;2―实验值
图 2 体膨胀温度曲线
Fig.2 Temperature dependence of volume thermal expansion
同,结果如图3所示。不过,随着温度升高,理论值与实验值的偏差随温度上升明显增大。相对第一原理的结果,德拜-格林奈森模型的膨胀系数整体上显示出更大的偏差,仅在室温以下与实验值较接近。
图4和图5所示分别为线膨胀与线膨胀系数的理论曲线,其中实线与虚线分别为a轴和c轴分量,未能找到相应的实验资料。理论计算显示出hcp结构金属Ti的热膨胀有一定程度的各向异性。由于理论计算是先得出任一温度下的平衡晶格常数a和c(即先得出线膨胀),再由a和c得出相应的体积(即前述体膨胀是由线膨胀结果算出),故将体膨胀与实验结果相比较,可以认为在相应的温度范围内,线膨胀的理论值是可信的。

1―第一原理理论曲线;2―德拜-格林奈森理论曲线;
3―实验值
图 3 体膨胀系数温度曲线
Fig.3 Temperature dependence of coefficient of volume thermal expansion

1―εa;2―εc
图 4 线膨胀温度曲线
Fig.4 Temperature dependence of linear thermal expansion

1―αa;2―αc
图 5 线膨胀系数温度曲线
Fig.5 Temperature dependence of coefficient of linear thermal expansion
由0 K时平衡晶格常数下得到的声子谱,从式(4)求出定容摩尔热容CV。根据线热膨胀的计算结果,从0 K到1 000 K,每100 K取1组平衡晶格常数,共11组,求出相应晶格常数下的弹性常数,由弹性常数得到体弹性模量,经样条插值,给出任一温度下的体弹性模量,再由式(5)即得到定压摩尔热容。图6中实线与圆点分别为定压摩尔热容Cp理论曲线与实验值,两者基本相符。作为对比,图中还给出对应0 K晶格常数的晶格定容摩尔热容 ,其中,双划线为根据声子谱由式(4)得出的结果,虚线为德拜理论计算值,德拜温度为432 K。
,其中,双划线为根据声子谱由式(4)得出的结果,虚线为德拜理论计算值,德拜温度为432 K。

1―Cp;2―(第一原理);3―CV(德拜理论);4―实验值
图 6 摩尔热容温度曲线
Fig.6 Temperature dependence of molar heat capacities
从图6可看出,高温时,晶格定容摩尔热容趋于常数3R(R为摩尔气体常数),这与理论结果相符;而定压摩尔热容实验值则随温度呈近似线性增加,这与式(5)计算结果相符。根据式(5),高温时,Cp近似与温度T成比例。从图6还可看出第一原理计算值与德拜理论值的差别。德拜理论值在低温的极限是严格正确的,这是因为此时只有低频振动模才对热容有贡献。在高温时,两者都给出定容热容的经典极限3R。只有在中温区域,两者才显示出差别,而且差别不大,这也说明德拜热容理论能用于计算晶格定容热容。
3 讨 论
对比热膨胀ε与热膨胀系数α的理论与实验偏差,可以发现前者小于后者。或者说,可以较为准确地计算不同温度下的晶格常数,但对热膨胀系数的计算与实验偏差相对较大。这是因为晶格常数的改变近似与热膨胀系数和温度改变的乘积成正比,考虑到ε与α的数量级分别为10-3与10-6,即使α与实验的偏差较大,在不太大的温区内,由此产生的ε偏差也是较小的。另外,热膨胀系数的计算用到数值导数,对原始数据的噪声非常敏感。因而在前面的计算中ε与实验较符合,而α则偏差较大。在高温区较大的偏差也有误差的累积效应,LDA近似低估晶格常数并且高估弹性模量,这样将低估热膨胀系数,进一步低估晶格常数且高估弹性模量,这种效应累积使得在高温区热膨胀系数的偏差越来越大。另外,在高温区,热膨胀系数理论值与实验值的偏差较大,且随着温度升高较快地增加,可能表明了准谐近似的失效。准谐近似只是考虑了部分非谐效应。对于大多数材料,非谐效应比较小,但当温度升高时,非谐效应会变得比较重要。对于Ti,在1 155 K将发生hcp→bcc相变,可以预料有很强的非谐效应。在准谐近似下,非谐效应对晶格振动频率的影响表现在振动频率随晶格参数变化,但在特定晶格参数下,振动频率与温度无关。实际上,由于非谐效应的影响,晶格振动频率也直接与温度有关。非谐效应使简谐近似下的振动频率发生偏移,在高温区,振动频率偏移量与温度近似成正比,且与频率有关。
根据德拜-格林奈森模型热膨胀系数也可等价地表示为格林奈森关系, 。在此模型中,格林奈森参数γ为常数,体弹性模量B为基态(T=0 K)晶格参数下理论值,也是常数。这就导致膨胀系数α与温度的关系完全由晶格振动热容CV决定,因而,在高温区,α将随着CV趋于常数。实际上,在一般情况下,格林奈森参数γ将随温度增大而增大,而体弹性模量B随温度增大而减小,因而热膨胀系数将随温度持续增加。在德拜-格林奈森模型中,体弹性模量B为0 K晶格参数值下求得,格林奈森参数γ也是根据基态附近德拜温度的变化求出,这就决定了由此计算出的热膨胀系数只在T=0 K附近较为准确。Moruzzi等[4]报道了由此方案计算的立方结构材料的热膨胀系数在T=300 K以下与实验符合得较好。为此提出了一些改进方案[6],使得在德拜-格林奈森模型下的计算得到改进。由于采用了一些近似假定,这些方案下的计算精度仍然比第一原理的计算精度低。特别是这些改进方案很难推广到非立方结构材料,因而基于密度泛函微扰理论计算非立方结构材料热膨胀系数,是一种较好的方案。
。在此模型中,格林奈森参数γ为常数,体弹性模量B为基态(T=0 K)晶格参数下理论值,也是常数。这就导致膨胀系数α与温度的关系完全由晶格振动热容CV决定,因而,在高温区,α将随着CV趋于常数。实际上,在一般情况下,格林奈森参数γ将随温度增大而增大,而体弹性模量B随温度增大而减小,因而热膨胀系数将随温度持续增加。在德拜-格林奈森模型中,体弹性模量B为0 K晶格参数值下求得,格林奈森参数γ也是根据基态附近德拜温度的变化求出,这就决定了由此计算出的热膨胀系数只在T=0 K附近较为准确。Moruzzi等[4]报道了由此方案计算的立方结构材料的热膨胀系数在T=300 K以下与实验符合得较好。为此提出了一些改进方案[6],使得在德拜-格林奈森模型下的计算得到改进。由于采用了一些近似假定,这些方案下的计算精度仍然比第一原理的计算精度低。特别是这些改进方案很难推广到非立方结构材料,因而基于密度泛函微扰理论计算非立方结构材料热膨胀系数,是一种较好的方案。
由式(9)得出γa=1.38和γc=1.54。根据式(8),各向异性热膨胀起因于非谐振动的各向异性与弹性的各向异性。非谐振动的各向异性由格林奈森参数描述;弹性的各向异性可由比值γ=(S13+S33)/(S11+S12)表征。作为比较,对于立方系材料,该比值为λ=1;接近理想轴比的hcp结构Mg,γ=1.04,Mg有几乎各向同性的热膨胀。对Ti,γ=0.98,弹性的各向异性很小,但它在c方向的非谐振动明显大于a方向的非谐振动,因而Ti在c方向的热膨胀更大。
4 结 论
a. 在295 K时hcp金属Ti声子谱理论曲线与实验值在[001]方向上的光学纵模有少许偏差,其余部分较相符。
b. Ti显示出各向异性热膨胀,在c轴与a轴方向热膨胀系数比约为1.5。Ti的热膨胀各向异性是由其非谐振动的各向异性决定的。体膨胀与体膨胀系数计算结果在较宽的温度范围内与已有的实验数据相符。随着温度增加,误差迅速增大,这一方面是误差的累积效应所致,另一方面表明接近hcp→bcc相变点,非谐效应迅速增强,因而准谐近似失效。
c. 基于德拜-格林奈森模型德热膨胀系数计算值在低温区与实验值相符,但由于计算中所用的参量都由温度为0 K附近晶格参数决定,因而在高温区理论值与实验值有显著的偏差。
d. 晶格定容摩尔热容的第一原理计算值与德拜理论值在中温区有少许差别,在低温区与高温区非常接近。德拜热容理论可以很好地描述晶格定容热容。定压摩尔热容的理论计算值与实验值在较宽的温度范围与实验相符。
e. 运用基于密度泛函与密度泛函微扰理论的第一原理方法可以较准确地计算各向异性晶体的热学 性质。
参考文献:
[1] 李燕峰, 徐 慧, 宋招权, 等. Zn掺杂对MgB2电子结构及超导转变温度的影响[J]. 中南大学学报: 自然科学版, 2006, 37(5): 925-931.
LI Yan-feng, XU Hui, SONG Zhao-quan, et al. Effect of Zn doping on the electronic structure and superconductivity temperature of MgB2[J]. J Cent South Univ: Science and Technology, 2006, 37(5): 925-931.
[2] 孙 坚, 姚 强. 相弹性性质和堆垛层错能的第一原理计算[J]. 中国有色金属学报, 2006, 16(7): 1166-1170.
SUN Jian, YAO Qiang. First-principles study of elastic properties and stacking fault energies of ZrCr2 Laves phase[J]. The Chinese Journal of Nonferrous Metals, 2006, 16(7): 1166-1170.
[3] 陶辉锦, 谢佑卿, 彭红建, 等. 面心立方和亚稳液相Cu的原子状态及物理性质与温度变化关系的Debye-Grüneisen模型研究[J]. 中南大学学报: 自然科学版, 2006, 37(6): 1036-1042.
TAO Hui-jing, XIE You-qing, PENG Hong-jian, et al. Debye-Grüneisen model study of the temperature dependence of physical properties of FCC and metastable Cu[J]. J Cent South Univ: Science and Technology, 2006, 37(6): 1036-1042.
[4] Moruzzi V L, Janak J F, Schwarz K. Calculated thermal properties of metals[J]. Phys Rev B, 1988, 37(2): 790-799.
[5] JIN Hong-mei, WU Ping. First-principles calculation of thermal coefficient Part 1. Cubic metals[J]. Journal of Alloys and Compounds, 2002, 343(1/2): 71-76.
[6] LU Xiao-gang, Selleby M, Sundman B. Theoretical modeling of molar volume and thermal[J]. Acta Material, 2005, 53: 2259-2272.
[7] Mayera B, Antona H, Botta E, et al. Ab initio calculation of the elastic constants and thermal expansion coefficients of Laves phases[J]. Intermetallics, 2003, 11(12): 23-32.
[8] Baroni S, Giannozzi P, Andrea T. Green’s-function approach to linear response in solids[J]. Phys Rev Lett, 1987, 58(16): 1861-1864.
[9] Giannozzi P, de Gironcoli S, Pavone P, et al. Ab initio calculation of phonon dispersions in semiconductors[J]. Phys Rev B, 1991, 43(9): 7231-7242.
[10] Narasimham S, de Gironcoli S. Ab initio calculation of the thermal properties of Cu: Performance of the LDA and GGA[J]. Phys Rev B, 2002, 65(6): 064302(1-7).
[11] Quong A A, Liu A Y. First-principles calculations of the thermal expansion of metals[J]. Phys Rev B, 1997, 56(13): 7767-7770.
[12] XIE Jian-jun, de Gironcoli S, Baroni S, et al. First-principles calculation of the thermal properties of silver[J]. Phys Rev B, 1999, 59(2): 965-969.
[13] Debernardi A, Alouani M, Dreysse H. Ab initio thermodynamics of metals: Al and W[J]. Phys Rev B, 2001, 63(6): 064305(1-7).
[14] Wang S Q. First-principles study of the anisotropic thermal expansion of wurtzite ZnS[J]. Appl Phys Lett, 2006, 88(6): 061902(1-3).
[15] Gonze X, Beuken J M, Caracas R, et al. First-principles computation of material properties: The abinit software project[J]. Computational Materials Science, 2002, 25: 478-492.
[16] Troullier N, Martins J L. Efficient pseudopotentials for plane-wave calculations[J]. Phys Rev B, 1991, 43(3): 1993-2006.
[17] Gray D E. American institute of physics handbook[M]. New York: McGaw-Hill Book Company, 1972.
[18] Barrera G D, Bruno J A O, Barron T H K, et al. Negative thermal expansion[J]. J Phys: Condens Matter, 2005, 17: R217-R251.
[19] Stassis C, Arch D, Harmon B N, et al. Lattice dynamics of hcp Ti[J]. Phys Rev B, 1979, 19(1): 181-188.
收稿日期:2007-02-22;修回日期:2007-04-26
基金项目:国家自然科学基金资助项目(50271085)
作者简介:聂耀庄(1965-),男,湖南岳阳人,博士研究生,讲师,从事计算材料及计算物理研究
通信作者:聂耀庄,男,博士研究生;电话: 0731-8660059;E-mail: yz_nie@163.com

