稀有金属 2007,(04),430-433 DOI:10.13373/j.cnki.cjrm.2007.04.025
钨合金强化机制的电子结构表征
李云凯 李玉秋 尹静静
北京理工大学材料科学与工程学院,北京理工大学材料科学与工程学院,北京理工大学材料科学与工程学院,北京理工大学材料科学与工程学院 北京100081,北京100081,北京100081,北京100081
摘 要:
结合固体与分子经验电子理论 (EET) 和改进的TFD理论计算了不同成分钨合金的相空间及相界面处的价电子结构。结果表明, 钨合金的强化机制与粘结相的价电子结构有着密切的关系。相对于纯钨, 粘结相的共价电子个数有所增加, 相界面处的电子密度和界面上可能的原子状态组数也大大提高, 且降低了钨颗粒之间的连接, 这是钨合金强化的本质原因。
关键词:
钨合金 ;强化 ;价电子结构 ;
中图分类号: TF125.241
作者简介: 李云凯 (E-mail:liyunkai@bit.edu.cn) ;
收稿日期: 2006-12-15
基金: 总装备部预研基金资助项目;
Electronic Structure Characterization for Strengthening of Tungsten Alloy
Abstract:
According to empirical electron theory of solid and molecules and the modified TFD theory, the valence electron structures of phase space and interface in different component tungsten alloys were calculated.The result showed that there was a closed relation between electron structure of cementation phase and strengthening effect on tungsten alloy.The cementation phase increased the electron density on the interphase, reduced the joining between tungsten grains.Cementation phase can increase the tensile strength.
Keyword:
tungsten alloy;strengthener;valence electron structures;
Received: 2006-12-15
高比重钨合金是一种以钨为主, 加入少量Ni, Fe, Mn或Cu等元素组成的合金, 通常是通过液相烧结制备而成。 合金的微观组织是单质的钨颗粒分布在粘结相内, 钨颗粒是一种硬而脆的相, 而基体是一种韧性相。 这类合金具有一系列优异的物理机械性能, 如强度高、 硬度高、 延性好、 韧性好、 热膨胀系数小、 导电导热性好、 抗腐蚀和抗氧化性好、 机加工和可焊性好等, 因此在国防军工、 航空航天、 电子信息、 能源、 冶金等领域中具有十分广泛的用途, 在国民经济中占有重要地位
[1 ,2 ,3 ]
。 众所周知, 在钨合金中, 粘结相是一个重要的组成部分, 它的加入改善了材料的烧结性能, 提高了材料的强度。 以往的研究主要致力于裂纹的萌生、 扩展及断裂方面, 但这仅停留在宏观上, 要想进一步揭示粘结相对其强度塑型的影响, 还必须从微观的角度去探究。
20世纪70年代末, 基于价键理论
[4 ]
和能带理论建立的固体与分子经验电子理论 (EET)
[5 ,6 ]
, 提供了一个处理复杂体系价电子结构的简捷使用的经验方法――键距差 (BLD) 法, 使得研究合金的宏观性能可以追溯到合金原子的价电子结构层次, 为合金改性设计提供了深层次的理论指导
[7 ,8 ,9 ,10 ]
。 程氏改进的TFD理论
[11 ]
提出, 电子的运动状态决定了材料特性, 并认为固体中各原子相互接触的表面上电子密度必须连续。 本文以EET的原子杂化状态为基础, 通过计算不同粘结相的钨合金中各相的电子密度来阐述粘结相的增强机制。
1 固溶体价电子结构计算的基本思想
1.1 平均原子模型
在对无序二元合金固溶体进行价电子结构计算时, 文献
[
5 ]
曾分别对代位固溶体和间隙固溶体提出了平均原子模型和平均晶胞模型。 平均原子模型的基本思想是将溶剂原子A与溶质原子B组成的晶体设想为由A和B混合而成的单一“原子”S组成, 这种原子的特征参数n C , n L , R (1) 等为相应的A和B原子的特征参数的计权平均值, 由此按BLD方法计算出的价电子结构即为固溶体的价电子结构。
1.2 点阵参数的计算方法
由EET的键距差分析方法计算合金相的价电子结构必须已知晶体结构及晶格常数, 这给计算带来了一定的困难。 文献
[
12 ]
中提出, 虽然含溶质晶胞的点阵参数不能直接从实验中获得, 但由于单键半距是表征原子尺度的特征量, 且形成共价键的两个原子间的距离可以看作是两个原子的单键半距之和, 基于金属学中点阵常数与原子半径成正比这一事实, 可以认为代位固溶体中含溶质原子晶胞的点阵常数与溶剂、 溶质原子单键半距之和成正比, 即:
a =η {R A (1) +R B (1) } (1)
式中, R A (1) 为固溶体中溶剂原子的单键半距; R B (1) 为溶质原子的单键半距; a 为含溶质晶胞的点阵常数; η 为一系数。 从式 (1) 中可以看出, 当溶剂中不含溶质原子时, B原子占据的位置仍为A原子所占据, 因此, 可以借助这一“边界条件”确定系数η , 此时R B (1) =R A (1) =R A0 (1) , a =a 0 , R A0 为纯A原子晶体时A原子的单键半距; a 0 为纯溶剂原子组成的晶体的点阵参数, 所以
η =a 0 /2R A0 (1) (2)
将式 (2) 代入式 (1) 得
a =a 0 {R A (1) +R B (1) }/2R A0 (1) (3)
1.3 电子密度的计算
相界面处的电子密度ρ , 电子密度的差值Δρ , 使电子密度保持连续的原子状态组数σ N 以及Δρ >10%或Δρ <10%时, 界面上可能的原子状态组数σ N ′都是决定相界面处结合强度的重要参数。 相界面处电子密度ρ 越高, 界面结合越强; 相界面处的电子密度差Δρ 越小, 界面上的电子密度连续性越好, 界面应力越小, 反之界面应力越大。 同样的, 界面上可能的原子状态组数越多, 界面的连续性就越好。
相界面处电子密度等于界面上的共价电子数与界面面积之比, 即
ρ = ∑ n C S , 电子密度差的计算公式为:
Δ ρ = | ρ 1 - ρ 2 | [ ρ 1 + ρ 2 ] × 1 2 × 1 0 0 % [ 1 1 ] 。
2 钨合金中的价电子结构及界面处结合因子计算
现在分别以镍锰比为8∶2, 6∶4, 4∶6的钨镍锰 (90 W) 合金和镍铁比为7∶3的钨镍铁 (93 W) 合金为例, 来计算钨合金的价电子结构。
在这里, 认为钨镍锰固溶体由纯钨晶胞、 镍锰晶胞以及含钨的镍锰晶胞混合而成
[8 ]
。 其中, 纯钨晶胞为BCC型结构, 晶格常数为0.316524 nm。 根据BLD分析计算结果
[5 ,13 ]
, 取钨位于甲种杂化第三阶。
镍锰晶胞是含有锰的镍晶胞, 根据平均原子模型的思想, 将溶剂原子Ni和溶质原子Mn组成的晶体设想为由Ni和Mn混合而成的单一“原子”S, 由此按BLD方法计算出的价电子结构即为镍锰固溶体的价电子结构, 同理可得镍铁固溶体的价电子结构。 由3式可得, 镍锰比为8∶2, 6∶4, 4∶6, 镍铁比为7∶3晶胞的晶格常数分别为0.35722, 0.35722, 0.357271, 0.347495。
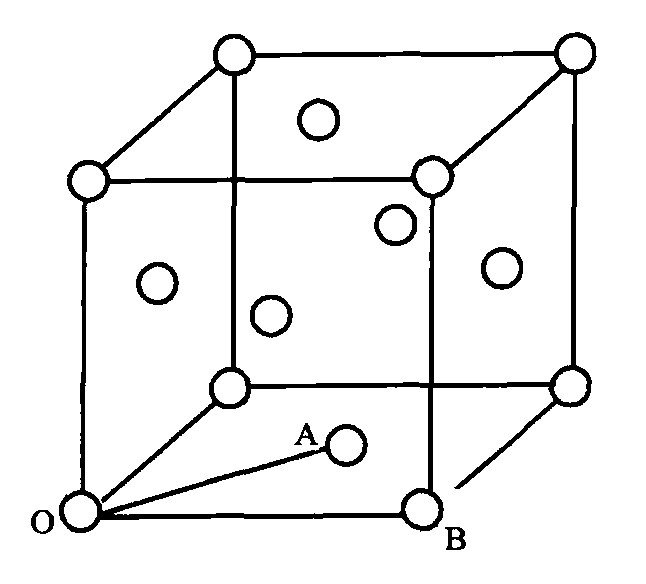
这样的镍锰和镍铁晶胞均为面心立方结构, 如图1所示。 取顶角处的O原子为参考原子, 它与处于面心位置的A原子形成A键, 与角上的B原子形成B键。 在分子和晶体内, 由相同的原子形成的键距相同的各键成为等同键。 则OA键和OB键相应的等同键数依据公式I α =I M I S I K
[5 ]
计算如下:
I A =12; I B =6
各实验键距为,
D n A = √ 2 2 a 0 , D n B = a 0 。 由BLD法就可求得各固溶体的价电子结构。 用同样的方法处理钨镍锰及钨镍铁合金, 将W原子和S原子组成的晶体设想为由W和S混合而成的单一“原子”S′, 按BLD方法计算出的价电子结构即为钨镍锰及钨镍铁固溶体的价电子结构。
W是体心立方结构, 取其密排面即 (110) 面为所计算的面, 具体计算方法参照文献
[
11 ]
。 假设在一个钨颗粒内部, 原子堆垛面之间的结合也服从刘志林等提出的界面规律, 则由于钨颗粒内部的电子状态均一致, 则结合面处的电子密度都相同, 电子密度差为0。 另外, 由于Ni-Mn, Ni-Fe, W-Ni-Mn, W-Ni-Fe均为面心立方结构, 因此取 (111) 面为所计算的面。 计算方法同上。
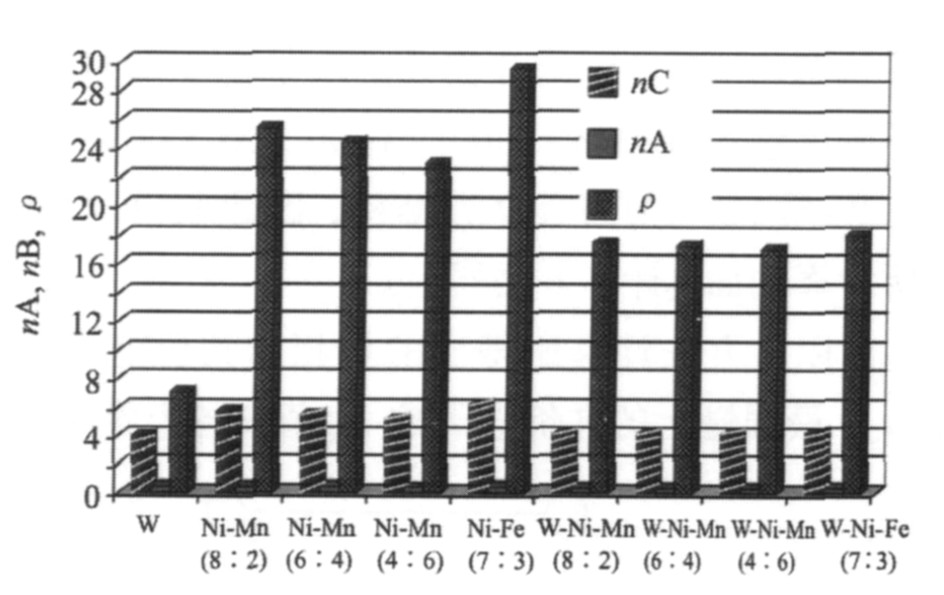
由以上方法计算所得出的各相的共价电子总数n C 、 最强键上的共价电子数n A 值和各个界面上的电子密度ρ 、 电子密度差Δρ 以及可能的原子状态组数σ N ′值列入表1和2中。 为了便于比较, 将表中的数据表示在图2中。
图1 Ni-Mn/Ni-Fe合金晶胞
Fig.1 Cell of Ni-Mn/Ni-Fe alloy
表1 W, 粘结相和固溶体的nC, nA以及ρ值
Table 1 Values of n C n A and ρ in W, cementation phases and solid solutions
Elements
n C n A ρ
W
4
0.43579
7.057825
Ni-Mn (8∶2)
5.668502
0.468153
25.41773791
Ni-Mn (6∶4)
5.437812
0.449101
24.38333517
Ni-Mn (4∶6)
5.130955
0.423760
23.00091153
Ni-Fe (7∶3)
6.225862
0.513654
29.47095254
W-Ni-Mn (8∶2)
4.16685
0.344551
17.41859157
W-Ni-Mn (6∶4)
4.143781
0.342669
17.24389008
W-Ni-Mn (4∶6)
4.113096
0.340175
16.98222843
W-Ni-Fe (7∶3)
4.222586
0.346497
17.97755876
表2W/W, W/固溶体以及固溶体/粘结相界面上的Δρ和σN′值
Table 2 Values of Δρ and σ N on W/W, W/solid solutions, solid solutions/cementation phases
Elements
Δρ /%
σ N ′
W/W
0
1
W/W-Ni-Mn (8∶2)
84.6591782
1
W/W-Ni-Mn (6∶4)
83.8300099
1
W/W-Ni-Mn (4∶6)
82.5655687
1
W/W-Ni-Fe (7∶3)
87.2344028
1
Ni-Mn/W-Ni-Mn (8∶2)
37.3474872
128
Ni-Mn/W-Ni-Mn (6∶4)
34.3018068
83
Ni-Mn/W-Ni-Mn (4∶6)
30.1061052
52
Ni-Mn/W-Ni-Fe (7∶3)
48.4457508
114
图2 W, 粘结相以及固溶体的nC, nA以及ρ值比较
Fig.2 Contrast figure of values of n C , n A and ρ ′s between W, cementation phases and solid solutions
3 结果与讨论
一 般认为钨合金的断裂模式有4种: (1) 钨颗粒-钨颗粒界面分离; (2) 钨颗粒-粘结相界面分离; (3) 粘结相的韧断; (4) 钨颗粒的解理断裂。 由文献
[
8 ]
可知, W-W界面的数量及W-粘结相结合力的大小是影响钨合金力学性能的重要因素。 其中, W-W界面越多, 材料强度和韧性就越差, W-粘结相结合力越大则材料强度越高。 EET理论提出, 价电子个数是原子间结合的主要基础, 其中共价电子对数与强度密切相关, 共价电子对数越多, 则原子间结合越强。 因此钨合金的断裂模式和断裂强度与其相空间的共价电子数目和界面结合因子ρ , Δρ 以及σ N ′密切相关, 即共价电子数越多, 强度越高; 界面上的ρ 越高, Δρ 越小, σ N ′越大, 则界面结合越好。 根据表1和图2可明显看出, 钨颗粒内部的n C 和n A 分别是4和0.43579, 而粘结相镍锰 (8∶2, 6∶4, 4∶6) 以及镍铁的n C 分别是5.668502, 5.437812, 5.130955和6.225862, n A 分别为0.468153, 0.449101, 0.423760以及0.513654。 显然粘结相的n C 和n A 值要高于纯钨。 尤其是Ni-Fe, n C 和n A 分别达到了6.225862和0.513654, 远远高出纯钨的4和0.43579。 因此, 粘结相的强度要高于纯钨。 W-W界面上存在大量缺陷, 与完整的W颗粒内部相比, 结合面上电子密度较低, 结合强度较低
[14 ,15 ,16 ]
, 一旦钨颗粒间直接接触而形成W-W界面, 则此界面在变形中优先形成裂纹源, 所以W-W界面是钨合金中的弱相。 钨合金中的粘结相分布在钨颗粒之间, 降低了钨颗粒的连接度, 提高了材料的强 度。
钨 合金中存在着两种界面, 即W-W界面、 W-粘结相界面。 其中W-粘结相界面上由于存在着钨与粘结相的互相固溶而产生了固溶体, 因此这里实际上出现了两种界面, 即W-固溶体界面, 固溶体-粘结相界面。 从表1, 2可得, 在钨颗粒内部, 解理面上的电子密度是7.057825, 电子密度差Δρ 为0, 但σ N 仅为1。 添加粘结相之后, 一级近似下界面上电子密度是偏离连续的, 但界面上的电子密度大大增加, W-Ni-Mn和W-Ni-Fe上的电子密度分别提高到了17.41859175, 17.24389008, 16.98222843, 17.97755876, σ N ′均维持在1。 从表中还可以看出, W-固溶体界面 (即W/W-Ni-Mn (8∶2, 6∶4, 4∶6) 与W/W-Ni-Fe) 上的电子密度差分别为84.6591782%, 83.8300099%, 82.5655687%, 87.2344028%, 均大于10%, 一级近似下都是不连续的, 界面偏离连续抑制了钨颗粒的长大。 可以看出, Ni-Mn (8∶2, 6∶4, 4∶6) 与Ni-Fe的电子密度和界面处的σ N ′有明显的提高。 其中电子密度分别提高到了25.41773791, 24.38333517, 23.00091153, 29.47095254, σ N ′则提高到128, 83, 52, 114。 总体上来说, 提高了界面的连续性, 改善了界面性能。
因此加入粘结相之后, 一方面降低了钨颗粒的连接度, 引入了强度较高的粘结相; 一方面提高了界面处的电子密度, 增加了界面上可能的原子状态组数, 提高了界面的连续性, 从而对钨合金起到增强作用。
4 结 论
1. 钨合金的强化机理与粘结相的价电子结构有着密切的关系。
2. 钨合金中粘结相 (尤其是Ni-Fe) 的共价电子总数n C 和最强键上的共价电子对数n A 高出纯钨, 强度较纯钨高, 且分布在钨颗粒之间, 减低了钨颗粒的连接度, 起到增强作用。
3. 添加粘结相之后, 提高了界面处的电子密度, 增加了界面上可能的原子状态组数, 提高了界面的连续性。 钨合金的断裂方式由钨颗粒界面断裂转变为钨颗粒的解理断裂。
参考文献
[1] 叶途明, 易健宏, 彭元东, 胡礼福, 吕豫湘.纳米晶高密度钨合金的研究进展[J].稀有金属, 2004, 28 (4) :726.
[2] 叶途明, 易健宏, 李丽娅, 彭元东, 吕豫湘, 胡礼福.高比重钨合金研究的新进展[J].材料导报, 2003, 17 (12) :15.
[3] 沈宏芳, 陈文革, 谷臣清.纳米W-Ni-Fe高比重合金的研究[J].稀有金属, 2005, 29 (2) :133.
[4] 鲍林L著.化学键本质[M].上海:上海科学技术出版社, 1966.1.
[5] 张瑞林.固体与分子经验电子理论[M].长春:吉林科学技术出版社, 1993.1.
[6] 陈松, 刘泽光, 陈登权, 罗锡明, 许昆, 邓德国.Au/Sn界面互扩散特征[J].稀有金属, 2005, 29 (4) :413.
[7] 刘志林, 孙振国, 李志林.余氏理论和程氏理论在合金研究中的应用[J].自然科学进展, 1998, 8 (2) :150.
[8] 刘志林.合金价电子结构与成分设计[M].长春:吉林科学技术出版社, 1990.1.
[9] 解峰.高密度合金W-Ni-Mn液相烧结[J].中国稀土学报, 2005, 23 (12) :168.
[10] 屈华, 刘伟东, 张坤, 刘志林.Ti3Al基合金 (0001) (α2) // (110) β界面价电子结构分析[J].稀有金属材料与工程, 2005, (10) :61.
[11] 刘志林, 李志林, 刘伟东.界面电子结构与界面性能[M].北京:科学出版社, 2002.1.
[12] 孙振国, 瞿晓剑, 顾琳.固溶体的价电子结构与固溶强化[J].材料科学与工艺, 2005, 13 (5) :475.
[13] 高英俊, 钟夏平, 刘慧, 班冬梅.Al-Cu合金亚稳相的价电子结构分析[J].稀有金属, 2003, 27 (6) :846.
[14] 许宝才, 李淑华, 叶明惠, 赵忠民.高比重钨合金的烧接成形与组织性能研究[J].军械工程学院学报, 2004, 16 (5) :67.
[15] 刘海燕, 李智芳, 宁建国.91钨合金断裂行为研究[J].太原理工大学学报, 2005, 36 (6) :717.
[16] 王国栋, 郭让民, 冯宝奇, 赵鸿磊, 刘建章.W-Ni-Fe断裂行为与断口形貌分析[J].稀有金属, 2005, 29 (1) :115.