文章编号:1004-0609(2009)10-1759-07
铝晶体自由表面的稳定性计算
张新明,刘建才,唐建国,陈明安
(中南大学 材料科学与工程学院 有色金属材料科学与工程教育部重点实验室,长沙 410083)
摘 要:采用第一原理赝势平面波方法,计算铝晶体学自由表面(100)、(110)和(111)的能量、几何与电子结构。根据表面能计算预测铝自由表面结构的稳定性。结果表明:铝自由表面结构稳定性由强到弱的顺序为(111)、(100)、(110);表面原子驰豫不仅引起表面几何结构的变化,而且使表面层的电子结构与键合特性发生改变;(100)、(110)和(111)表层驰豫分别为3.337%、-6.147%和-2.364%;表面电荷密度不同引起表面能差异,表面原子层和次表面原子层的电荷面密度在s和p轨道上重新分布;表面原子层电荷密度越大,表面能越低。
关键词:Al晶体;第一原理;表面驰豫;表面能
中图分类号:TG146.1 文献标识码:A
Calculation of stability of free surfaces in aluminum crystal
ZHANG Xin-ming, LIU Jian-cai, TANG Jian-guo, CHEN Ming-an
(Key Laboratory of Non-ferrous Metal Materials Science and Engineering, Ministry of Education, School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: The surface energy, atomic geometry and electronic structures of Al(100), (110) and (111) free surfaces were calculated using the method of supercell and the first-principles pseudopotential plane waves within generalized gradient approximation. According to the calculated surface energy, the structural stability of Al free surfaces from strong to weak is predicted in the order as (111), (100) and (110). The relaxation of the surface atom layers not only causes the change of geometrical structures of the surface models, but also leads to the variation of their electronic structures and bonding characters. For the (100), (110) and (111) free surfaces, the calculated surfaces relaxation are 3.337%, -6.147% and -2.364%, respectively. The surface energy is related to the surface electron density distribution, the electron density of orbital s and p of the first two surface atom layers redistributes. The higher the surface electron density is, the lower the surface energy is.
Key words: aluminum crystal; first principle; surface relaxation; surface energy
铝电解电容器是电子器件的重要元件之一,在电子信息设备、仪器、机电和家电等产品中有着不可替代的重要作用。随着电子器件的小型化发展,必然要求电解电容器电容量更大,体积更小[1]。人们通过探索和研究发现,借助电化学的方法,沿铝箔{100}晶体面腐蚀出大量的隧道孔,可以大幅度提高铝箔的表面积和比电容[2]。因此,人们一直采取各种措施增加高纯铝箔中立方织构{100}<001>的占有率,使之超过95%[3-4]。人们虽然致力于提高高压铝箔{100}的占有率,但在铝箔中不仅存在{100}平行于轧面的结构,而且存在其它类型的晶体学平面结构,如{110}和{111}等。不同的晶体学平面对于腐蚀过程具有不同影响[5],因此,对于铝箔不同晶体学平面的活性的研究显得非常重要。
虽然现在已经发展了很多测定表面原子结构的实验方法[6]:毛细管法、临界表面张力法和估算法等,但测量值仅对液体比较准确,对固体则有很大的局限性,测量结果往往因测试试样与测试条件的不一致而存在一定差异。同时,由于晶体表面具有各向异性,实验方法很难区分是哪个晶体表面的表面能。所以通过理论的方法来获得表面能意义巨大。
近年来,很多学者采用不同的计算方法对不同的晶体表面结构进行了计算[7-9],但对于Al晶体表面仍缺乏系统与全面的研究[10-13],且这些结果大多侧重于表面能与表面相对面间角的关系,而很少考虑表面驰豫对结构的影响。本文作者采用第一原理的计算方法,对铝箔中常见的3个晶体学平面{100}、{110}和{111}的构型、驰豫、表面电子结构及表面能进行了比较系统的研究,该结果对了解铝箔表面性质及进一步揭示铝箔表面不同晶面在腐蚀过程中的作用提供了参考。
1 不同晶体学表面结构稳定性的热力学分析
表面能可以理解为晶体在外力作用下晶体解理断裂所需的能量,其大小取决于表面原子间的相互作用,与表面原子的几何结构密切相关。对于无掺杂的晶体,可采用热力学-准化学方法[14]来初步判断其表面结构稳定性。
若一个晶体包含NA个原子,每个原子有Z个配位原子,则共有ZNA/2个结合键,假如每个结合键的键能为εAA,则晶体的升华热?HS就是破断所有键所需能量,即

对于某个特定表面,若表面原子与体内原子相比少了n个结合键,则这n个键就属于表面,其表面结合能?U(即表面能)可估计如下:

式中:NS为单位表面上的原子数。FCC晶体体内每个原子的最近邻原子数为N=12,若形成(111)表面,则表面原子丧失n=3个结合键,即存在3个“悬挂键”。 (111)面相应的表面能?U(111)和单位表面上的原子数 如下:
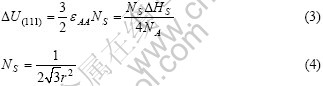
同理可得,对于(110)面:

对于(110)面:

因此,这3个特征晶体面的表面能比值如下:

从这个定性的结果可知,虽然从上面的表达式中不能直接计算出FCC晶体平面的表面能:但由此却可以对其相对值的大小进行比较。结果显示,各晶面的表面能由大到小顺序为(100)、(110)、(111),(100)面的表面活性最高,(111)面的表面稳定性最好。
热力学理论认为,晶体总是趋向于以密排面作为外表面,以达到最稳定的结构。铝为面心密排结构,(111)是最密排面,(111)面的原子面密度是14.1 nm-2;(100)面的原子面密度是12.3 nm-2,(110)面的原子面密度是8.7 nm-2。由原子面密度可以认为各个晶体表面能由大到小顺序为:(110)、(100)、(111),与前面用化学估算法不符。这是因为准化学方法只是一个定性的结论。对于不同种类的原子,由于其原子结构,尤其是其电子结构存在差异,表面原子的几何和电子结构会发生变化,计算的表面能值会有所不同。此外,在上面的计算中,其表面能的计算仅仅近似地计算了其“化学项”的作用,而没有考虑表面弛豫与重构等“结构项”的影响。为了进一步揭示Al晶体不同表面的结构稳定性与表面几何和电子结构的关系,本文作者对Al晶体的表面结构进行了深入分析。
2 计算模型与方法
计算采用基于密度泛函理论的赝势平面波方法-CASTEP(Cambridge serial total energy package)软件包完成[15]。采用梯度近似(GGA)的交换关联(PBE)的超软赝势(Ultrasoft)来描述离子实与价电子之间的相互作用(3s23p1),结构模型采用BFGS算法进行优化与驰豫。计算中采用三维周期性边界条件的超晶胞模型模拟表面,为了保证z方向原子层不相互作用,必须要有足够厚的真空层,一般认为,真空厚度为10?以上能满足要求。由于在晶胞中,原子层数的多少是决定表面有效性的一个重要因素,通常认为5个原子层厚度足够。有效表面除了要求必要的原子层数之外,还要求有合适的初始固定层数,以保证表面层原子能够充分驰豫。通过原子层数和真空层厚度测试,将(100)、(110)和(111)表面模型的原子层都取7层。计算时固定晶胞中最底层的3层原子,晶胞模型见图1所示。在计算时动能截断点取350 eV,布里渊区积分采用Monkhorst-Pack形式的特殊K点方法,FFT网格取 (24×24×24)。对应Al(111)、 (110)和(100)表面模型,K点分别取(10×10×1)、(6×9×1)和(9×9×1)。自洽计算时,所有的总能量收敛值设为1.0×10-5 eV/atom,每个原子上的力小于0.03 eV/?,公差偏移小于0.001 ?,应力偏差小于0.05 GPa。
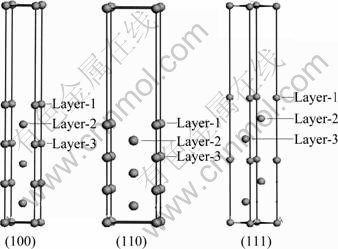
图1 铝(100)、(110)和(111)的表面模型
Fig.1 Surface models of (100), (110) and (111) of aluminum crystal
3 分析与讨论
3.1 体模型的测试
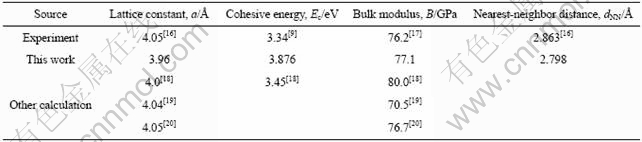
为了验证程序结果的可靠性,本文作者计算了由4个铝原子组成的FCC-Al晶体的晶格常数a、结合能Ec、体模量B和最近邻原子间距dNN,并与实验值及报道的计算结果进行了比较,结果如表1所列。结果表明,Al的晶格常数a、Ec、B和dNN与实验值均符合较好,说明计算模型可行。
表1 Al的晶格常数a、结合能Ec、体模量B和最近邻原子间距dNN
Table 1 Lattice constant a, cohesive energy Ec, bulk modulus B and nearest-neighbor distance dNN of atoms for Al crystal
3.2 表面原子的驰豫
让表面结构进行充分驰豫,以消除加在表面层原子上的应力,从而得到更加稳定的结构。由于铝晶体的表面原子在充分驰豫条件下,基本上不发生重构,所以本文作者主要是考察表面原子的弛豫现象。为了定量描述Al表面原子驰豫情况,定义如下:

表2所列为Al表面模型原子弛豫的几何结构参数变化,其中d12为表面层与第2层之间间距,d23为第2层与第3层的间距,d34为第3层与第4层的间距。
表2 Al表面模型的几何结构参数
Table 2 Geometric structural parameters of Al surface models
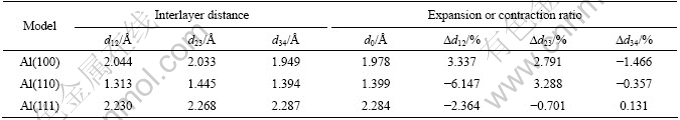
由此可以看出,3个不同晶面表面各相邻层之间存在不同程度的驰豫现象。相对FCC-Al晶体而言,(100)表面表层有轻微的向外伸展,而(110)和(111)表层都是向内收缩。Al(111)模型的第2层与第3层原子间的层间距仍然减少,即向内收缩;而Al(110)与Al(100)模型的第2层与第3层原子间的层间距增大,即向外膨胀。此外,从表2还可以看出|?d12|>|?d23|>|?d34|,即表面原子驰豫效应从表面第1层到第4层逐渐减弱,越靠内各相邻层之间距变化越小。Al(110)表面原子的弛豫程度差不多是Al(111)面和Al(100)面的2倍,而Al(100)表面又比Al(111)表面原子向内收缩程度大,表明Al(110)表面原子的驰豫程度最大,Al(100)表面次之,Al(111)表面最小,这说明Al(111)表面可能是较稳定的表面。
3.3 表面能及结构稳定性
表面能是一个重要的物理量,通过分析表面能,可以进一步了解表面稳定性和表面吸附能力等许多信息。这里只考虑基态(0 K)的情况,所以表面能的计算式定义如下:

由于本软件计算的是基态原子能量(p=0 Pa,T=0 K),因而pV和TS可以忽略不计,计算结果如表3所列。从表3可知,Al(111)表面能的计算结果比实验值略低一些,这主要是由于本计算的表面能没有考虑温度对表面的影响。计算得到的各个表面的表面能的大小顺序和分子动力学计算结果一致,其中Al(110)表面能最大,Al(100)表面能次之,Al(111)表面能最低,因而Al(111)表面最稳定,活性最差;Al(100)表面居中;Al(110)表面最不稳定、活性最高。值得指出的是,从第一原理计算所得的Al(111)、(100)与(110)的表面能变化趋势(即EAl(111)?EAl(100)?EAl(110)= 1?1.274?1.321)与采用热力学-准化学方法估算(即EAl(111)?EAl(100)?EAl(110)=1?1.115?1.021)的相比,虽然数值相差不大,但变化趋势却不完全相同。由热力学-准化学方法预计Al(100)表面的活性最高,而从第一原理的计算却表明表面活性最好的是Al (110)表面。第一原理计算结果顺序与表面原子密度顺序一致,表面原子密度越高,表面能越低,第一原理计算结果更加符合实际情况。
表3 Al晶体的表面参数
Table 3 Surface parameters of aluminum crystal
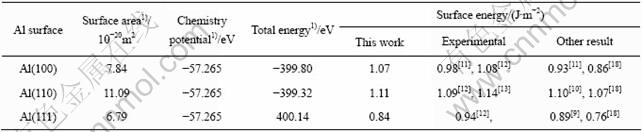 1) Results of this work.
1) Results of this work.
3.4 表面态密度和电子密度
为了进一步说明铝晶体不同表面的结构稳定性差异的电子机制,还计算了表面模型的电子总态密度和表面第一、二、三层的局域态密度(LDOS)及其分波态密度(PDOS)(见图2所示)。由图2可知,Al晶体中价电子态密度主要由3s和3p分波态密度构成,其中绝大部分由p电子贡献,费密能级(EF)附近存在明显的赝能隙。比较Al 3个表面与铝晶体密度分布可知,除Al(111)表面外其它形成表面模型的总态密度曲线相对Al晶体改变不大,但在Al晶体赝能隙区中约4 eV处表面模型却新出现了一微小的表面态,其中以Al(100)表面最明显,而Al(100)与Al(110)表面则还进一步削弱了费密能级处的价电子峰,使赝能隙区价电子密度整体抬高。显然,正是这些在高能级区新增的表面态与低能级区减少的电子态密度使表面结构处于比晶体结构能量相对较高的状态,因而出现了表面能,而其增减的相对幅度则决定了表面的相对稳定性与活性。实际上,这个结论也可从计算模型费密能级绝对值大小的比较中得到初步说明。Al晶体的EF=8.504 eV,Al(111)、 Al(110)和Al(100)表面的EF分别为3.005、1.306和2.401 eV。相对于晶体,表面的费密能级低,因此表面的稳定性比晶体低;而不同表面中,稳定性最好的是Al(111)面,Al(100)面次之,稳定性最差的则是Al(110)面。
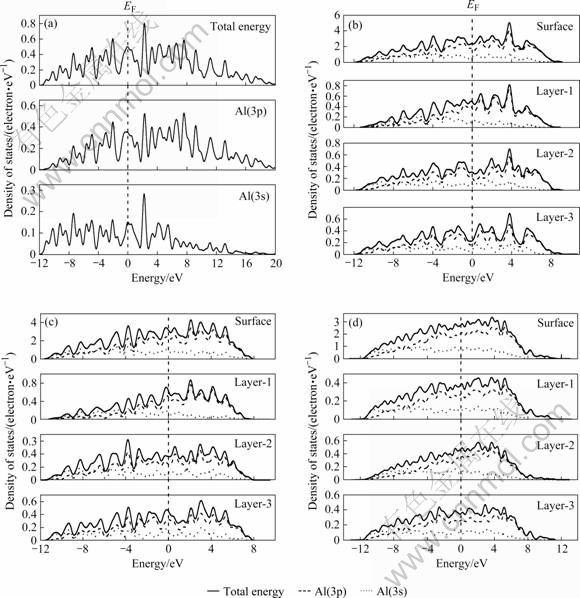
图2 Al晶体与表面的电子态密度图
Fig.2 Density of states of aluminum crystal and surfaces: (a) Al bulk; (b) Al(100); (c) Al(110); (d) Al(111)
进一步对Al表面不同原子层的局域电子态密度进行比较时发现:表面层原子(Layer-1)的局域电子态密度相对于与内层原子(Layer-3)发生了很大变化。表面原子层上电荷面密度Al(111)面最低,Al(110)面最高。显然,正是这些表面层电子态密度的变化引起了不同表面结构稳定性的差异。进一步比较不同原子层局域电子态密度不难看出,对表面电子结构造成影响的主要是Layer-1和Layer-2。比较Layer-2与Layer-3的局域电子态密度可见,Layer-2层的原子也处于较高的电子能态,因而具有较高的活化能力,因此,这也是热力学-准化学方法不能准确预测不同表面结构稳定性的一个原因。随着原子层远离表面而向体内深入,原子层的LDOS的体态特征越来越明显,同时表面态迅速衰减。
LDOS充分显示在局域态密度中,3p态密度占极重要的成分,且在各原子层的局域态密度中,3s电子均局域于价带的低能端,对费密能级富集的态密度几乎无贡献,因此,表面态几乎全部由3p态所贡献。另一方面,就3s态密度而言,第二原子层中的3s分波态密度就收敛成体态中的3s分波态密度,相比之下,局域的3p分波态密度向体态的3p分波态密度的收敛却慢得多,这意味这Al中的3p电子对Al表面的某些特性起主导作用。更细微地比较图中相对应的局域态密度及其分波态密度可以发现,3s态密度的分布几乎相同,局域态密度的差异主要体现在3p态,这说明,Al表面结构的驰豫主要与3p电子有关。
4 结论
1) Al晶体表面能由大到小顺序为(110)、(100)、(111);(111)面的结构稳定性最高,(100)面次之,(110)面的稳定性最差。即相同条件下,Al(110)表面活性最高,最易与其它物质反应,(111)活性最低最稳定。
2) Al(110)表面向内收缩程度约为是Al(111)面与Al(100)面的2倍。表面原子驰豫不仅引起表面几何结构的变化,还使表面层的电子结构与键合特性发生改变,使表面层原子的电子态密度峰型发生了变化,表面能值由表面下两层原子层的电荷面密度在s和p轨道上的分布决定。表面电荷面密度越高,表面能越低。
REFERENCES
[1] MAO W M, JIANG H, YANG P, FENG H P, YU Y N. Distribution of microelements and their influence on the corrosion behavior of aluminum foil[J]. Journal of Materials Science & Technology, 2005, 21(1): 43-46.
[2] XIAO Y Q, LIU C M, JIANG S, CHEN Z Y, ZHANG X M. Influence of high hot-rolling temperature and multistage annealing upon the cube texture of high purity aluminum capacitor foils[J]. Textures of Materials, 2002, 408/412(4): 1449-1452.
[3] ZHANG X M, LIU S D, TANG J G, ZHOU Z P. Mechanism of strengthening of cube texture for high purity aluminum foils by additional-annealing[J]. Trans Nonferrous Met Soc China, 2003, 13(3): 499-503.
[4] LIU C M, ZHANG X M, CHEN Z Y, DENG Y L, ZHOU Z P. Evolution of recrystallization textures in high voltage aluminum capacitor foils[J]. Trans Nonferrous Met Soc China, 2001, 11(4): 513-516.
[5] 杨 宏. 非铬酸系电解电容器用高压阳极铝箔的研究[D]. 北京: 北京科技大学, 2007.
YANG Hong. Research on the non-chromic acid high voltage anode aluminum foil for electrolytic capacitor[D]. Beijing: University of Science and Technology Beijing, 2007.
[6] RTYSON W, MW A. Surface free energies of solid metals: Estimation from liquid surface tension measurements[J]. Surf Sci, 1977, 62(1): 267-276.
[7] METHFESSEL M, HENNING D, SCHEZER M. Trends of the surface relaxations, surface energies, and work functions of the 4d transition metals[J]. Phys Rev B, 1992, 46(8): 4816-4829.
[8] KOLLAR J, VITOS L, SKRIVER H L. Surface energy and work function of the light actinides[J]. Phys Rev B, 1994, 49(16): 11288-11292.
[9] RODRIGUEZ A M, BOZZOLO G, FERRANTE J. Multilayer relaxation and surface energies of fcc and bcc metals using equivalent crystal theory[J]. Surf Sci, 1993, 289(1/2): 100-126.
[10] WANG X C, YU J, QIAN K Y, WANG F, MA J X, HU X. The calculation of the surface energy of high-index surfaces in metals at zero temperature[J]. Surface Science, 2004, 551(3): 179-188.
[11] MUTASA B, FARKAS D. Atomistic structure of high-index surfaces in metals and alloys[J]. Surface Science, 1998, 415(3): 312-319.
[12] SCHOCHLIN J, BOHNEN K P, HO K M. Structure and dynamical at the Al(111)-surface[J]. Surf Sci, 1995, 324(2/3): 113-121.
[13] RAEKER T J, DE PRISTO A E. Corrected effective-medium method. IV. Bulk cohesive and surface energies of second-and third-row metals and multilayer relaxation of Al, Fe, and Ni[J]. Phys Rev B, 1989, 39(14): 9967-9982.
[14] 肖纪美, 朱逢吾. 材料能量学[M]. 上海: 上海科学技术出版社, 1999: 417.
XIAO Ji-mei, ZHU Feng-wu. Material energetics[M]. Shanghai: Shanghai Science and Technology Press, 1999: 417.
[15] SEGALL M D, LINDAN P, PROBERT M J. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Condense Matter, 2002, 14(11): 2717-2744.
[16] STRAUMANIS M E, WOODARD C L. Lattice parameters and thermal expansion coefficients of Al, Ag and Mo at low temperatures[J]. Acta Cryst A, 1971, 27: 549-551.
[17] TALLON J L, WOLFENDEN A. Temperature dependence of the elastic constants of aluminum[J]. J Phys Chem Solids, 1979, 40(11): 831-837.
[18] 张芳英, 滕英元, 张美霞, 朱圣龙. Al(001)、Al(110)、Al(111)面表面能的密度泛函理论计算[J]. 腐蚀科学与防护技术, 2005, 17(1): 47-49.
ZHANG Fang-ying, TENG Ying-yuan, ZHANG Mei-xia, ZHU Sheng-long. Density functional theory study of surface energies of Al(001), (110) and (111)[J]. Corrosion Science and Protection Technology, 2005, 17(1): 47-49.
[19] KIEJNA A, LUNDQVIST B I. First-principles study of surface and subsurface O structures at Al(111)[J]. Physical Review B, 2001, 63(8): 085405(1-10).
[20] ZHUKOVSKII Y F, JACOBS P W M, CAUS? M. On the mechanism of the interaction between oxygen and close -packed single- crystal aluminum surfaces[J]. J Phys Chem Solids, 2003, 64(8): 1317-1331.
基金项目:国家重点基础研究发展计划资助项目(2005CB623706)
收稿日期:2008-10-27;修订日期:2009-01-03
通信作者:张新明,教授,博士;电话:0731-8830265;E-mial: jcliu1568@yahoo.com.cn
(编辑 龙怀中)

