Quantum chemical calculations on structure of Mo8O264-
来源期刊:中国有色金属学报(英文版)2002年第5期
论文作者:吴争平 尹周澜 陈启元 张平民
文章页码:1001 - 1006
Key words:quantum chemistry calculation; Mo8O264-; structure
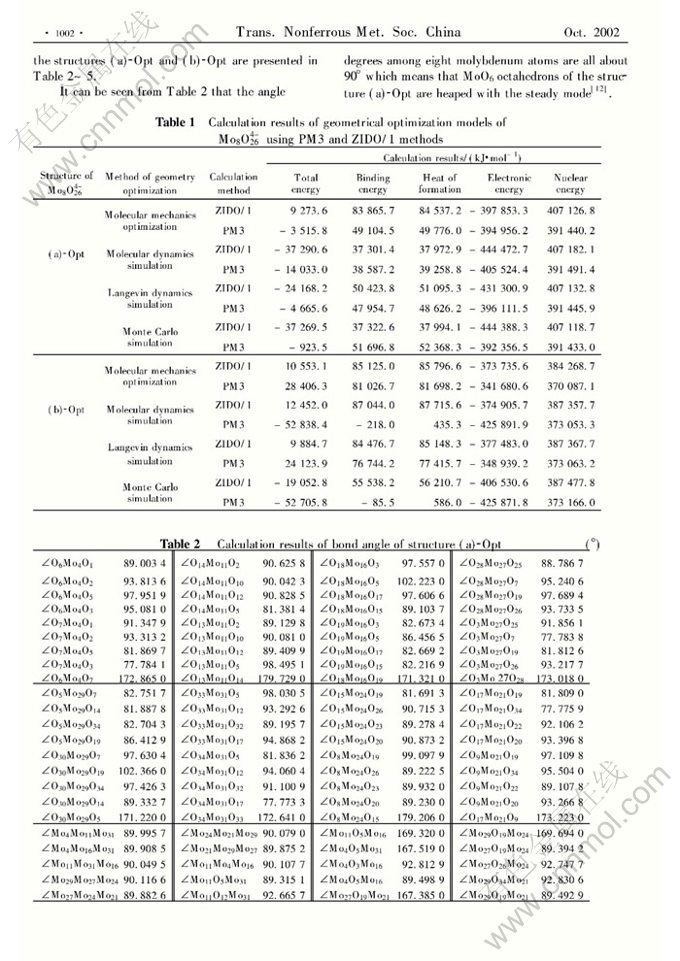
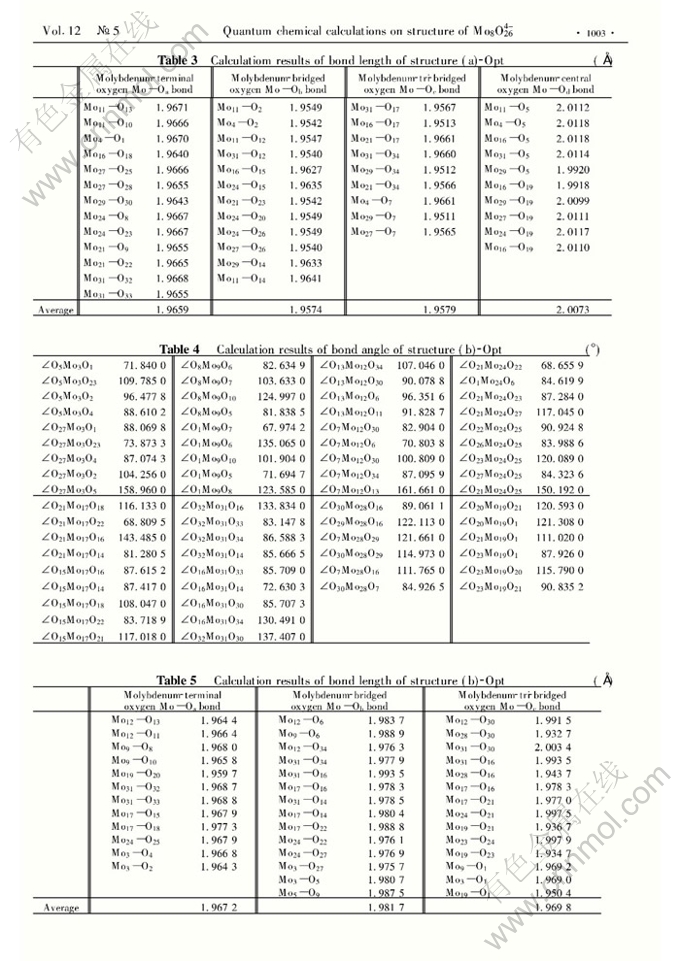
Abstract: Two possible structures of Mo8O264-) were optimized using the Polak-Ribiere method of Molecular Mechanics Optimization and the termination condition is RMS (Root-mean-square) gradient of 0.42kJ/mol. Based on the calculations of the molecular dynamics, Lengevin dynamics and Monte Carlo dynamics simulation, the structure models of Mo8O264- with the lowest energy were acquired respectively according to the energy of the systems calculated using the ZIDO/1 and PM3 methods. The total energy, energies of some frontier molecular orbitals and atomic charges of Mo8O264- were computed at the HF/3-21G and HF/STO-3G levels. The calculation results show that the contortion of the structure with eight MoO6 is smaller than that of the structure with six MoO6 and two MoO4. The total energies of the two structures are nearly equal because the contortion of the structure with six MoO6 and two MoO4 would make the exclusion force decreased.