
Potential energies of characteristic atoms on basis of experimental heats of formation of AuCu and AuCu3 compounds (Ⅱ)
XIE You-qing(谢佑卿)1, 2, 3, LI Xiao-bo(李晓波)4, LIU Xin-bi(刘心笔)1, 2, 3,
PENG Hong-jian(彭红建)1, 2, 3, NIE Yao-zhuang(聂耀庄)1, 2, 3
1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Powder Metallurgy Research Institute, Central South University, Changsha 410083, China;
3. State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China;
4. College of Mechanical Engineering, Xiangtan University, Xiangtan 411105, China
Received 30 March 2009; accepted 2 June 2009
_______________________________________________________________________________
Abstract: The potential energy sequences of characteristic atoms were separated out by nine potential energy E-functions on the basis of larger experimental heats of formation of the L10-AuCu and L12-AuCu3 compounds only. According to these potential energy sequences of characteristic atoms, the potential energies and heats of formation of disordered Au1-xCux alloys were calculated by corresponding E-functions at 0 K; and the potential energies, heats of formation and critical Tc-temperatures of the order-disorder transitions of L10-AuCu, L12-Au3Cu and L12-AuCu3 compounds, Au3Cu-, AuCu- and AuCu3-type ordered alloys with maximal ordering degrees were also calculated at 0 K. The results obtained by both the first and present parts of this investigation were compared. Comparing the results obtained by nine E-functions, the 5th E(x, 0, σ) function may be chosen for describing thermodynamic properties of the compounds, ordered and disordered phases and for establishing the phase diagram of the Au-Cu system in the future.
Key words: systematic science of alloys; Au-Cu system; potential energy; heat of formation; phase transition temperature
_______________________________________________________________________________
1 Introduction
In the energetic aspect, the systematic science of alloys(SSA) is a framework of the total energy able to be separated out. It can not only accurately describe phase regions in the phase diagram, but also precisely describe the variations in thermodynamic properties, such as general vibration heat capacity  general vibration energy Uv(x, T, σ), general vibration entropy Sv(x, T, σ), enthalpy H(x, T, σ), characteristic Gibbs energy G*(x, T, σ) without containing configuration entropy and Gibbs energy G(x, T, σ) containing configuration entropy Sc(x, σ) as functions of composition, temperature and ordering degree, within the phase regions. Therefore, it is very important to separate out potential energy sequences of characteristic atoms on the basis of accurate heats of formation of several compounds in the alloy system.
general vibration energy Uv(x, T, σ), general vibration entropy Sv(x, T, σ), enthalpy H(x, T, σ), characteristic Gibbs energy G*(x, T, σ) without containing configuration entropy and Gibbs energy G(x, T, σ) containing configuration entropy Sc(x, σ) as functions of composition, temperature and ordering degree, within the phase regions. Therefore, it is very important to separate out potential energy sequences of characteristic atoms on the basis of accurate heats of formation of several compounds in the alloy system.
In the first part of this investigation, the potential energy sequences of the  and
and  characteristic atoms in Au-Cu system were separated out respectively by nine E-functions on the basis of smaller experimental heats of formation of the AuCu and AuCu3 compounds: ?Hexp(AuCu, 298, 1)=-8 746 J/mol and ?Hexp(AuCu3, 298, 1)=-7 164 J/mol[1-2]. According to these potential energy sequences of the and characteristic atoms, the potential energies E(x, 0, 0) and heats of formation ?H(x, 0, 0) of disordered Au1-xCux alloys, the potential energies, heats of formation and order-disorder transition Tc-temperatures of Au3Cu, AuCu and AuCu3 compound as well as the Au3Cu-, AuCu- and AuCu3-type ordered alloys with maximal ordering degree were calculated. And finally, the 5th E-function was determined to be suitable for describing Au-Cu system.
characteristic atoms in Au-Cu system were separated out respectively by nine E-functions on the basis of smaller experimental heats of formation of the AuCu and AuCu3 compounds: ?Hexp(AuCu, 298, 1)=-8 746 J/mol and ?Hexp(AuCu3, 298, 1)=-7 164 J/mol[1-2]. According to these potential energy sequences of the and characteristic atoms, the potential energies E(x, 0, 0) and heats of formation ?H(x, 0, 0) of disordered Au1-xCux alloys, the potential energies, heats of formation and order-disorder transition Tc-temperatures of Au3Cu, AuCu and AuCu3 compound as well as the Au3Cu-, AuCu- and AuCu3-type ordered alloys with maximal ordering degree were calculated. And finally, the 5th E-function was determined to be suitable for describing Au-Cu system.
In the present part of this investigation, the potential energy sequences of the and characteristic atoms in Au-Cu system were separated out respectively by nine E-functions on the basis of larger experimental heats of formation of the AuCu and AuCu3 compounds: ?Hexp(AuCu, 298, 1)=-9 337 J/mol and ?Hexp(AuCu3, 298, 1)=-7 268 J/mol[3-4]. According to these potential energy sequences of the and characteristic atoms, the potential energies E(x, 0, 0), heats of formation ?H(x, 0, 0) of disordered Au1-xCux alloys, the potential energies, heats of formation and order-disorder transition Tc-temperatures of Au3Cu, AuCu and AuCu3 compound as well as the Au3Cu-, AuCu- and AuCu3-type ordered alloys with maximal ordering degree were calculated. And finally, the 5th E-function was still determined to be suitable for describing Au-Cu system.
2 Results
2.1 Potential energy sequences of characteristic atoms
On the basis of larger experimental heats of formation of the L10-AuCu and L12-AuCu3 compounds, the potential energy 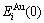 and
and 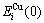 sequences of the and characteristic atoms in the Au-Cu system are separated out by nine E-functions, listed in Table 1 and shown in Fig.1, from which it can be known that the potential energy sequences obtained from nine E-functions are different each other, and different to ones obtained from the first part of this investigation.
sequences of the and characteristic atoms in the Au-Cu system are separated out by nine E-functions, listed in Table 1 and shown in Fig.1, from which it can be known that the potential energy sequences obtained from nine E-functions are different each other, and different to ones obtained from the first part of this investigation.
Table 1 Potential energy sequences of 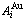 and
and  characteristic atoms in Au-Cu system calculated by nine E-functions at 0 K (J/mol)
characteristic atoms in Au-Cu system calculated by nine E-functions at 0 K (J/mol)


Fig.1 Potential energy  and
and  sequences of and characteristic atoms in Au-Cu system (
sequences of and characteristic atoms in Au-Cu system (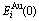 and
and  sequences obtained by the 4th (a), 5th (b) and 7th (c) E-functions are different, even average potential energies of compounds, ordered and disordered alloys calculated by them are, respectively, equivalent (see results of 2.2, 2.3 and 2.4 in the present section))
sequences obtained by the 4th (a), 5th (b) and 7th (c) E-functions are different, even average potential energies of compounds, ordered and disordered alloys calculated by them are, respectively, equivalent (see results of 2.2, 2.3 and 2.4 in the present section))
2.2 Average potential energies E(x, 0, 0), EAu(x, 0, 0) and ECu(x, 0, 0) of disordered Au1-xCux alloys and their components at 0 K
According to the potential energy sequences of the characteristic atoms, the E(x, 0, 0), EAu(x, 0, 0) and ECu(x, 0, 0) potential energies of disordered Au1-xCux alloys and their components as well as their ?H(x, 0, 0) heats of formation calculated by nine E-functions are listed in Table 2 and shown in Fig.2, from which the following knowledge can be obtained.
Table 2 Potential energies E(x, 0, 0), EAu(x, 0, 0), ECu(x, 0, 0) and heats of function ?H(x, 0, 0) of disordered Au1-xCux alloys calculated by nine E-functions at 0 K (J/mol)


Fig.2 Average potential energies E(x, 0, 0), EAu(x, 0, 0) and ECu(x, 0, 0) of disordered Au1-xCux alloys and their components calculated from the 2nd, 3rd, 4th, 5th and 7th E-functions (a); and heats of formation ?H(x, 0, 0) for disordered Au1-xCux alloys calculated from nine E-functions, together with experimental heats of formation at 320 K[4] (denoted by circles) (b)
1) The average E(x, 0, 0) potential energies of the disordered Au1-xCux alloys obtained from the nine functions are different each other. The average EAu(x, 0, 0) and ECu(x, 0, 0) potential energies of the Au and Cu components obtained by nine E-functions are different each other too. But there is no E-function, which can well describe the experimental heats of formation in the whole compositional range. It has been discovered that the experimental heats of formation of so-called disordered alloys are referred to partly-ordered alloys with short-range ordering(SRO) degrees[6-7]. The effect of ordering degrees on the heats of formation of the alloy phases are discussed in Section 3.
2) The heats of formation of the disordered Au1-xCux alloys obtained by the 5th E-function are higher than corresponding experimental values in the compositional range 25%≤x(Cu)≤75%. But it may be used to describe the Au-Cu system, if the experimental heats of formation of disordered alloys are referred to ones of partly-ordered alloys with SRO degrees.
2.3 Potential energies and heats of formation of L12- Au3Cu, L10-AuCu and L12-AuCu3 compounds
According to the potential energy sequences of the characteristic atoms in Table 1, the potential energies 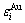 and
and  of characteristic atoms occupied at the ith site in the cell and the average potential energies, heats of formation and order-disorder transition temperatures of the L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds can be calculated by the additive law of properties of characteristic crystals and are listed in Table 3, from which the following knowledge can be obtained.
of characteristic atoms occupied at the ith site in the cell and the average potential energies, heats of formation and order-disorder transition temperatures of the L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds can be calculated by the additive law of properties of characteristic crystals and are listed in Table 3, from which the following knowledge can be obtained.
Table 3 Potential energies of characteristic atoms (ε, in 10-19 J/atom), total potential energies (E, in J/mol), heat of formation (?H, in J/mol) and order-disorder transformation temperatures (Tc, in K) of L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds at 0 K

1) The average potential energies of these compounds obtained from the 2nd to 9th E-functions are equal to each other, but their potential energies of characteristic atoms are different each other.
2) The average potential energies and heats of formation of the L10-AuCu and L12-AuCu3 compound obtained by the 5th E-function are equal to corresponding experimental values, and their Tc-temperatures are closer by experiment values than ones obtained by other E-functions.
3) The average potential energies and heats of formation of the L12-Au3Cu compound calculated from the 2nd to 9th E-functions are equal to each other and closer by experiment values, but only the Tc-temperature obtained by the 5th E-function is closer by the experiment value than ones obtained by other E-functions.
2.4 Energetic properties of ordered Au1-xCux alloys
According to the potential energy sequences and concentrations  and
and  in the ordered Au1-xCux alloys with ordering degree σ, their energetic properties can be calculated at 0 K. The average potential energies E(x, 0, σmax) and heats of formation ?H(x, 0, σmax) of the Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degree are listed in Tables 4-6. Their heats of formation ?H(x, 0, σmax) calculated by nine E-functions are shown in Fig.3.
in the ordered Au1-xCux alloys with ordering degree σ, their energetic properties can be calculated at 0 K. The average potential energies E(x, 0, σmax) and heats of formation ?H(x, 0, σmax) of the Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degree are listed in Tables 4-6. Their heats of formation ?H(x, 0, σmax) calculated by nine E-functions are shown in Fig.3.

Fig.3 Heats of formation ?H(x, 0, σmax) of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degree calculated by nine E-functions at 0 K (Experimental values denoted by circles are respectively: ?H(Au3Cu)=-5 736 J/mol, ?H(AuCu)=-9 337 J/mol, ?H(AuCu3)=-7 268 J/mol[3-4])
Table 4 Potential energies E(x, 0, σmax) and heats heats of formation ?H(x, 0, σmax) of Au3Cu-type ordered Au1-xCux alloys with maximal ordering degrees calculated by nine E-functions at 0 K (J/mol)
Table 5 Potential energies E(x, 0, σmax) and heats heats of formation ?H(x, 0, σmax) of AuCu-type ordered Au1-xCux alloys with maximal ordering degrees calculated by nine E-functions at 0 K (J/mol)

Table 6 Potential energies E(x, 0, σmax) and heats heats of formation ?H(x, 0, σmax) of AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees calculated by nine E-functions at 0 K (J/mol)
2.5 Tc(x, σmax) temperatures of order-disorder phase transformations
The potential energy differences, 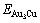 (x, 0, σmax)-Edis(x, 0, 0), EAuCu(x, 0, σmax)-Edis(x, 0, 0) and
(x, 0, σmax)-Edis(x, 0, 0), EAuCu(x, 0, σmax)-Edis(x, 0, 0) and 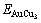 (x, 0, σmax)-Edis(x, 0, 0) between the ordered and disordered alloys, and the critical temperatures Tc(x, σmax) of Au3Cu-, AuCu-, and AuCu3-type Au1-xCux alloys translated into the disordered alloys described by nine E-functions have been calculated. The results are shown in Fig.4. The Tc-temperatures of these compounds described by the 5th E-function are closer by the experimental values than ones by other E-functions.
(x, 0, σmax)-Edis(x, 0, 0) between the ordered and disordered alloys, and the critical temperatures Tc(x, σmax) of Au3Cu-, AuCu-, and AuCu3-type Au1-xCux alloys translated into the disordered alloys described by nine E-functions have been calculated. The results are shown in Fig.4. The Tc-temperatures of these compounds described by the 5th E-function are closer by the experimental values than ones by other E-functions.

Fig.4 Potential energy differences of Au3Cu-, AuCu- and AuCu3-type ordered alloys with the maximum ordering degrees relative to disordered Au1-xCux alloys ((a), (b), (c)); critical temperatures of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys translated into disordered Au1-xCux alloys described by nine E-functions ((d), (e), (f)) together with experimental data[8] (denoted by triangles)
2.6 ΔHord(0)-ΔHdis(0)―Tc interrelated patterns
Here, we have made a comparison for the ΔHord(0)-ΔHdis(0)―Tc interrelated patterns of the AuCu, AuCu3 and Au3Cu compounds relative to corresponding disordered alloys obtained respectively in the first part and in the present part of this investigation. From Fig.5 the following knowledge can be obtained.
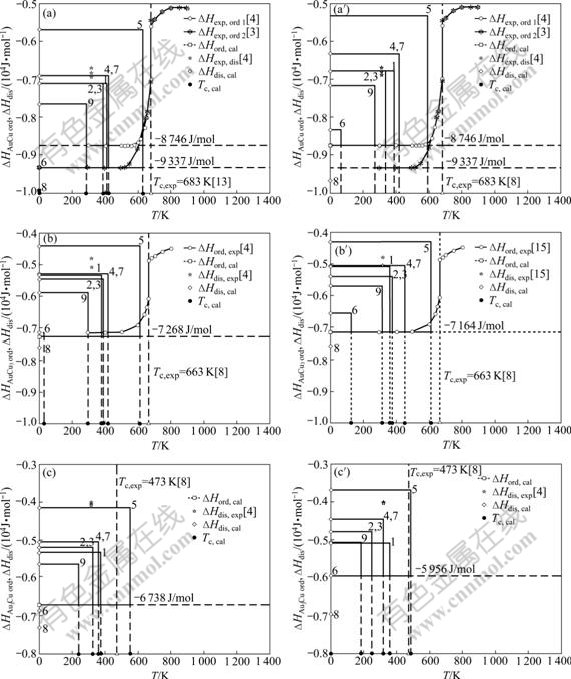
Fig.5 Comparison for ΔHord(0)-ΔHdis(0)―Tc interrelated patterns of AuCu, AuCu3 and Au3Cu compounds relative to corresponding disordered alloys obtained respectively from nine E-functions in present part ((a), (b), (c)) and the first part ((a′), (b′), (c′)) of this investigation
1) For the AuCu compound, although ?Hord(AuCu, 0, 1)=-9 337 J/mol of the AuCu compound obtained from the 2nd-9th E-functions in the present part are equivalent each other, ?Hdis(AuCu, 0, 0)=-5 690 J/mol obtained from the 5th E-function is higher (smallest absolute value) than that obtained from other E-functions. Therefore, the order-disorder transition temperature Tc(AuCu)=633 K obtained from the 5th E-function is the highest (see in Fig.5(a)). These results are, respectively, larger than ?Hord(AuCu, 0, 1)=-8 746 J/mol, ?Hdis(AuCu, 0, 0)=-5 330 J/mol and Tc(AuCu)= 593 K obtained in the first part of this investigation (see in Fig.5(a′)).
2) For the AuCu3 compound, there are analogous results to ones of the AuCu compounds. Although the ?Hord(AuCu3, 0, 1)=-7 268 J/mol obtained from the 2nd-9th E-functions in the present part are equivalent each other, ?Hdis(AuCu3, 0, 0)=-4 404 J/mol and Tc(AuCu3)=613 K obtained from the 5th E-function are the highest (see in Fig.5(b)). These results are, respectively, larger than ?Hord(AuCu3, 0, 1)=-7 164 J/mol, ?Hdis(AuCu3, 0, 0)=-4 309 J/mol and Tc(AuCu3)= 611 K obtained from the 5th E-function in the first part of this investigation (see in Fig.5(b′)).
3) For the Au3Cu compound, there are analogous results to ones for the AuCu and AuCu3 compounds. Although ?Hord(Au3Cu, 0, 1)=-6 738 J/mol obtained from the 2nd-9th E-functions in the present part are equivalent each other, the ?Hdis(Au3Cu, 0, 0)=-6 738 J/mol and Tc(Au3Cu)=558 K obtained from the 5th E-function are the highest (see in Fig.5(c)). These results are, respectively, larger than ?Hord(Au3Cu, 0, 1)=-5 956 J/mol, ?Hdis(Au3Cu, 0, 0)=-3 686 J/mol and Tc(Au3Cu)= 485 K obtained from the 5th E-function in the first part of this investigation (see in Fig.5(c′)).
3 Discussion on effect of ordering degree on heat of formation
In Au-Cu system containing ordered phases the effective interaction bond energy between the different kinds of atoms is bigger than one between the same kind of atoms, it leads to a tendency of atoms to be surrounded by atoms of the other kind and to a deviation of the distribution of concentration  and
and  of characteristic atoms and in a real alloy from the distribution in the completely disordered state. If this influence is long-range(LR) order, the original lattice splits into sublattices and each is occupied by only one kind of atom and the additional superstructure lines appear in Bragg scattering. If this influence is short-range(SR) order, it only affects the probability of finding an atom of a specific kind in the neighboring coordination shells around a certain atom, i.e., the concentration and of characteristic atoms and in a certain alloy. Therefore, the LR order degree σLR describes not only the deviation of the concentration and of characteristic atoms and in the LR ordered alloy from one in the disordered alloy, but also the relationship between atoms occupying right and wrong sublattices. But the SR order degree
of characteristic atoms and in a real alloy from the distribution in the completely disordered state. If this influence is long-range(LR) order, the original lattice splits into sublattices and each is occupied by only one kind of atom and the additional superstructure lines appear in Bragg scattering. If this influence is short-range(SR) order, it only affects the probability of finding an atom of a specific kind in the neighboring coordination shells around a certain atom, i.e., the concentration and of characteristic atoms and in a certain alloy. Therefore, the LR order degree σLR describes not only the deviation of the concentration and of characteristic atoms and in the LR ordered alloy from one in the disordered alloy, but also the relationship between atoms occupying right and wrong sublattices. But the SR order degree  describes only the deviation of the concentration of characteristic atoms in the SR ordered alloy from one in the disordered alloy.
describes only the deviation of the concentration of characteristic atoms in the SR ordered alloy from one in the disordered alloy.
Above critical temperatures, the stoichiometric AuCu and AuCu3 are statisfically face-centered cubic and a SR order exists. The experimental measurements of the SR order and the evaluation of X-ray data were carried out by ROBERTS[9], KEATING and WARREN [10]. The results are concluded as follows: 1) For the AuCu samples held at 698 K and 778 K above the critical temperature 683 K, there is high SR order degree, as evidenced by the first neighbor parameters which are respectively 37% and 36% of the way from complete disorder to perfect order. 2) The SR order degree for the quenched AuCu sample form 773 K is considerably higher than that for the sample held at this temperature, as evidenced by the first neighbor parameter which is 47.4% of the way from complete disorder to perfect order, indicating that the quenching of a large single crystal is inadequate to suppers increase in SR order degree. These results suggest that it is more difficult to get completely disordered alloy than to get fully ordered alloy in an alloy system containing ordered phases, that also has been demonstrated by the recent investigations [9-11].
Whether the heats of formation of disordered Au1-xCux alloys calculated from the 5th E-function on the basis of the smaller or larger heats of formation of the L10-AuCu and L12-AuCu3 compounds are higher than the experimental ones. This means that the so-called disordered Au1-xCux alloys contain SRO degrees.
In the SSA framework, the ordering of disordered alloys is the degeneracy of the energetic states of the characteristic atoms, and the disordering of ordered alloys is the splitting of the energetic states of the characteristic atoms. In this significance, both the LRO degree and the SRO degree can be described by the probabilities of the characteristic atoms occupied at the sublattices. This means that there are the same probabilities of characteristic atoms in the LRO and SRO alloys, if the σLRO=σSRO.
The effects of ordering degrees on the heats of formation ?H(x, 320, σ) of the Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys described by the 5th E-function in the first part and the present part of this investigation are shown in Fig.6, from which it can be known that the experimental heats of formation of the so-called disordered Au1-xCux alloys with the SRO degree should be described by three ?H(x, T, σ) functions
of the Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys in the three compositional ranges.
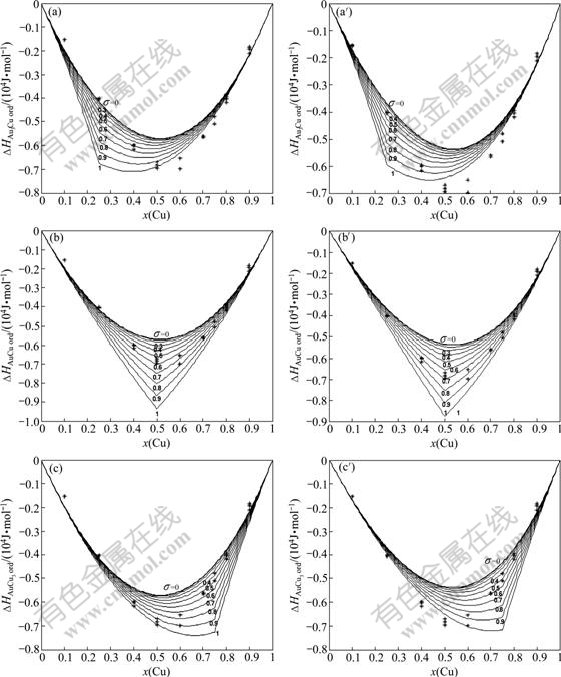
Fig.6 Effect of ordering degree on heats of formation of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys described by the 5th E-function in present part ((a), (b), (c)) and the first part ((a′), (b′), (c′)) of this investigation together with experimental data at 320 K [4]
In the present part of this investigation, the SRO degrees of the experimental Au1-xCux disordered alloys described by the 5th ?H(x, T, σ) function are shown in Figs.6(a), (b) and (c). For examples,

In the first part of this investigation, the SRO degrees of the experimental Au1-xCux disordered alloys described by the 5th ?H(x, T, σ) function are shown in Figs.6(a′), (b′) and (c′). For examples,

From this discussion, it can be concluded that it is more difficult to get completely disordered alloys than to get fully ordered alloys in an alloy system containing ordered alloy phases.
4 Conclusions
1) Based on the larger experimental heats of formation of the L10-AuCu and L12-AuCu3 compounds, the potential energy sequences for nine E-functions have been determined. Although the potential energies E(x, 0, 0) of the L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds calculated from these E-functions are, respectively, equivalent each other, the potential energies of their characteristic atoms are different each other.
2) The Tc-temperatures of the Au3Cu, AuCu and AuCu3 compounds obtained from the 5th E-function are closer by the experimental values than from other E-functions. The Tc-temperatures of the three compounds obtained from the 4th and 7th E-functions are greatly lower than experimental ones.
3) In an alloy system containing ordered alloy phases, it is more difficult to get completely disordered alloys than to get fully ordered alloys. The experimental heats of formation of the so-called disordered Au1-xCux alloys with SRO degree should be described by three ?H(x, T, σ)-functions of the Au3Cu-, AuCu- and AuCu3- type ordered Au1-xCux alloys in the three compositional ranges.
4) The ΔHord(0)-ΔHdis(0)―Tc interrelated patterns of the AuCu, AuCu3 and Au3Cu compounds relative to corresponding disordered alloys obtained respectively from nine E-functions in the present part and the first part of this investigation are compared. The potential energy sequences determined from the 5th E-function in both the first and the present parts may be chosen and developed into the energetic information chain. It will be necessary to make further investigation for understanding effects of potential energy sequences obtained on the basis of both smaller and larger experimental heats of formations on full thermodynamic properties within the phase regions and of phase transformations.
References
[1] XIE You-qing, LI Xiao-bo. LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang. Potential energies of characteristic atoms on basis of experimental heats of formation of AuCu and AuCu3 compounds (I) [J]. Trans Nonferrous Met Soc China, 2009, 19(5): 1243-1256.
[2] HULTGREN R, DESAI P D, HAWKINS D T, GLEISER M, KELLEY K K. Selected values of thermodynamic properties of binary metals and alloys [M]. Metal Park, OH: American Society for Metals; 1973.
[3] ORR R L, LUCIAT-LABRY J, HULTGREN R. Energy of the order-disorder transformation in Au-Cu [J]. Acta Metallurgica, 1960, 8: 431-434.
[4] ORR R L. Heats of formation of solid Au-Cu alloys [J]. Acta Metallurgica, 1960, 8: 489-493.
[5] KITTEL C. Solid state physics [M]. 6th ed. Wiley: New York, 1995.
[6] van TENDELOO G, AMELINCKS S, JENG S J, WAYMAN C M. The initial stages of ordering in CuAu (I) and CuAu (II) [J]. J Mater Sci, 1986, 21: 4395-4402.
[7] BATTEZZATI L, BELOTTI M, BRUNELLA V. Calorimetry of ordering and disordering in AuCu alloys [J]. Scripta Mater, 2001, 44: 2759-2764.
[8] BARRETT C S, MASSALSKI T B. Structure of metals [M]. 3rd ed. New York: McGraw, 1966.
[9] XIE You-qing, LI Xiao-bo, LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang. Potential energy sequences of characteristic atoms on basis of heats of formation of disordered Au1-xCux alloy (I) [J]. Journal of Materials Science and Engineering, 2009, 3: 51-68.
[10] KEATING D T, WARREN B E. Long-range in Beta-brass and Au3Cu [J]. J Applied Physis, 1951, 22: 286-290.
[11] XIE You-qing, LI Xiao-bo, LIU Xin-bi, LI Xiao-bo, PENG Hong-jian, NIE Yao-zhuang. Potential energy sequences of characteristic atoms on basis of heats of formation of disordered Au1-xCux alloys (II) [J]. Journal of Materials Science and Engineering, 2009, 6: 44-57.
_______________________________
Foundation item: Project(50471058) supported by the National Natural Science Foundation of China; Project(08JJ3099) supported by the Natural Science Foundation of Hunan Province, China
Corresponding author: XIE you-qing; Tel: +86-731-88879287; E-mail: xieyouq2000@yahoo.com.cn
DOI: 10.1016/S1003-6326(08)60437-9
(Edited by YANG Hua)

