АыУГІоКҫЙЁГиБҝИИ·ЁЖАјЫҝЙИИҙҰАнЗҝ»ҜВБәПҪрөДОціцДЈДвІОКэ
АҙФҙЖЪҝҜЈәЦР№ъУРЙ«ҪрКфС§ұЁ(УўОД°ж)2014ДкөЪ7ЖЪ
ВЫОДЧчХЯЈәAhmad FALAHATI Jun WU Peter LANG Mohammad Reza AHMADI Erwin POVODEN-KARADENIZ Ernst KOZESCHNIK
ОДХВТіВлЈә2157 - 2167
№ШјьҙКЈәІоКҫЙЁГиБҝИИЈ»ВБәПҪрЈ»Оціц¶ҜБҰС§Ј»ДЈДвЈ»ҝХО»Ј»MatCalc
Key wordsЈәdifferential scanning calorimetry; aluminum alloys; precipitation kinetics; simulation; vacancy; MatCalc
ХӘ ТӘЈәАыУГІоКҫЙЁГиБҝИИ·Ё(DSC)СРҫҝІ»Н¬өД№ММ¬Паұд·ҙУҰЎЈФЪМШ¶ЁөДИИҙҰАнЦЬЖЪЦРDSCРЕәЕәНПаөДЙъіЙУлИЬҪвХэПа№ШЎЈФЪ»щУЪКұјдәНОВ¶ИИИҙҰАнЦЬЖЪЦРЈ¬DSCјмІвөҪөДКФСщөДИИСЭұдҪб№ы¶ФУЪ·ЦОцПаСЭұдәНОціц¶ҜБҰС§ј«ОӘЦШТӘЎЈАыУГИИ/¶ҜБҰС§ДЈДвФӨІвБЛ6xxxәН2xxxВБәПҪрөДИИСЭұдDSCРЕәЕЎЈҙЛДЈРНН¬КұҝјВЗБЛІ»Н¬СЗОИПаЎўОИ¶ЁПаЎўҙг»рІРУаҝХО»өДЧҙМ¬әНУ°ПмЎЈАыУГНёЙдөзҫөәНёЯ·ЦұжВКөзҫөСйЦӨDSCЗъПЯЙПМШ¶Ё·еЦөөгОціцПаөДРОіЙЎўіЯҙзәНГЬ¶ИЎЈ
Abstract: Differential scanning calorimetry (DSC) has been used extensively to study different solid state reactions. The signals measured in DSC are associated with the growth and dissolution of different precipitates during a specific heat cycle. The time-temperature dependence of heat cycles and the corresponding heat flow evolution measured in the sample by DSC provide valuable experimental information about the phase evolution and the precipitation kinetics in the material. The thermo-kinetic computer simulation was used to predict the DSC signals of samples taken from 6xxx and 2xxx alloys. In the model, the evolution of different metastable and stable phases and the role and influence of excess quenched-in vacancies in the early stage of precipitation were taken into account. Transmission electron microscopy (TEM) and high-resolution TEM were used to verify the existence of precipitates, their size and number density at specific points of the DSC curves.
Trans. Nonferrous Met. Soc. China 24(2014) 2157-2167
Ahmad FALAHATI1, Jun WU1, Peter LANG2, Mohammad Reza AHMADI1, Erwin POVODEN-KARADENIZ3, Ernst KOZESCHNIK1,3
1. Institute of Materials Science and Technology, Vienna University of Technology,  9-11, 1040 Vienna, Austria;
9-11, 1040 Vienna, Austria;
2. Materials Center Leoben Forschung GmbH,  12, 8700 Leoben, Austria;
12, 8700 Leoben, Austria;
3. Christian Doppler Laboratory for Early Stages of Precipitation, Institute of Materials Science and Technology, Vienna University of Technology, 9-11, 1040 Vienna, Austria
Received 17 October 2013; accepted 30 April 2014
Abstract: Differential scanning calorimetry (DSC) has been used extensively to study different solid state reactions. The signals measured in DSC are associated with the growth and dissolution of different precipitates during a specific heat cycle. The time-temperature dependence of heat cycles and the corresponding heat flow evolution measured in the sample by DSC provide valuable experimental information about the phase evolution and the precipitation kinetics in the material. The thermo-kinetic computer simulation was used to predict the DSC signals of samples taken from 6xxx and 2xxx alloys. In the model, the evolution of different metastable and stable phases and the role and influence of excess quenched-in vacancies in the early stage of precipitation were taken into account. Transmission electron microscopy (TEM) and high-resolution TEM were used to verify the existence of precipitates, their size and number density at specific points of the DSC curves.
Key words: differential scanning calorimetry; aluminum alloys; precipitation kinetics; simulation; vacancy; MatCalc
1 Introduction
Understanding and simulating properly the precipitation kinetics during heat treatment of aluminum alloys are critical for achieving optimal materials microstructures. Over the last decade, with the increase of computational power, availability of thermodynamic databases especially for metastable phases, which depend on ab initio calculations [1] and existence of numerical solutions, we have the fundamental requirements to achieve reliable simulation of complex heat-treatment in aluminum alloys [2].
Before attempting to simulate complex heat treatment or calculation of their mechanical properties, a basic prerequisite is to get correct input parameters and to fine-tune the non-equilibrium kinetic parameters set for that specific material. A good methodology is to perform a non-isothermal continuous heating differential scanning calorimetry (DSC) experiment and to compare the experimental result with its DSC simulation curve and to obtain the required set of precipitation kinetic parameters. To get a reliable parameter set, at least verified qualitative knowledge about the phases, which exist at different important points on DSC curve, is necessary. Using a non-isothermal DSC has two important advantages. First, from experimental point of view, it is an effective, easy to use and rapid tool to measure with good precision for the energy absorbed (endothermic peaks) or released (exothermic peaks) during phase transformation by a well-defined small sample. Second, the energy absorbed or released is the overall effect of the evolution of precipitates and the composition of Al-matrix. DSC simulation is directly correlated to the phase fraction of metastable and stable phases and the state of the solid solution in the multi-component matrix as a function of time.
In the present work, a computational tool, MatCalc [3], for thermo-kinetic simulation of phase transforma- tion based on CALPHAD-type multicomponent thermodynamics is applied. Simulated DSC curves are calculated numerically as the first derivative of the system enthalpy with respect to temperature as given by
 (1)
(1)
where Q is the heat flow;  and
and  are the specific heat capacities of the alloy and pure reference aluminum, respectively; Hsys and HAl are the specific enthalpies of the alloy and pure Al, respectively; ЎчT/Ўчt is the heating rate.
are the specific heat capacities of the alloy and pure reference aluminum, respectively; Hsys and HAl are the specific enthalpies of the alloy and pure Al, respectively; ЎчT/Ўчt is the heating rate.
A proper physically-based predictive precipitation kinetics model approach can deliver a correct DSC simulation and as a result a correct precipitation kinetic parameter set, the same parameters set can be used for complex heat treatments and/or mechanical properties calculation. In the following, two industrial heat treatable aluminum alloys types 6xxx (Al-Mg-Si) and 2xxx (Al-Cu-Mg-Si) are chosen for demonstration of the simulation method and explaining important parameters in the precipitation kinetics simulation that should be taken into account. We assume that the reader has the fundamental knowledge about the experimental aspects of DSC [4].
2 Thermo-kinetic computational analysis
MatCalc version 5.52 (rel. 0.031) was used with the corresponding thermodynamic [5], mobility and physical databases [6] for Al alloys. Thermodynamics and mobility databases are CALPHAD-types. The thermodynamic database contains solid solution phases as a function of temperature and chemical composition and optimized Gibbs energy functions of stable and metastable compounds as a function of temperature [1]. The mobility database contains assessed self-diffusion and impurity diffusion data. The physical database contains optimized polynomials that describe the densities of phases as a function of temperature. Our simulation is based on multicomponent solid-state nucleation, which has been developed on the basis of classical nucleation theory (CNT). According to CNT, transient nucleation rate depends on the exponential expression of critical nucleation energy [7]. Individual nucleation events occur on a stochastic nature, but once a supercritical nucleus has formed, the deterministic laws of particle growth, dissolution and coarsening govern its further evolution.
In MatCalc, a mean-field approach to derive evolution equations for the growth and dissolution of spherical precipitates in multicomponent systems, which is based on the thermodynamic extremal principle (TEP), is used [8]. The numerical method implemented to follow up precipitates evolution (nucleation, growth and coarsening) is the numerical KampmannЁCWagner (NKW) model. The key quantity when evaluating precipitate nucleation rates in kinetic simulations model is the Gibbs free energy change of forming a critical nucleus. The total energy change due to nucleus formation, ЎчGnucl, can be separated into the following three contributions:

 (2)
(2)
where ҰӨGbulk is the chemical bulk free energy;  is the coherency elastic strain energy; ҰӨGsurf is the total interfacial free energy; Chemical bulk free energy (ҰӨGbulk) is associated with structural change from matrix to the precipitate phase; ҰС is the radius of the precipitate and
is the coherency elastic strain energy; ҰӨGsurf is the total interfacial free energy; Chemical bulk free energy (ҰӨGbulk) is associated with structural change from matrix to the precipitate phase; ҰС is the radius of the precipitate and  is the specific bulk energy, which is evaluated from the assessed CALPHAD-type free energies of the matrix and precipitate phases.
is the specific bulk energy, which is evaluated from the assessed CALPHAD-type free energies of the matrix and precipitate phases.
Nucleus formation is generally accompanied by a typical mismatch between the lattice parameters of the precipitate and the surrounding matrix. For a completely coherent interface, the strain energy () plays a crucial role since it is proportional to the total volume, whereas the interfacial energy is only proportional to the area of the nucleus. A simplified model for a solid spherical inclusion inserted into a spherical hole of an isotropic elastic matrix is a classic elasticity problem and the energy contribution coming from volumetric misfit stress is given by
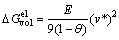 (3)
(3)
where E is the elastic modulus; ҰИ is Poisson ratio; v* is the misfit volume strain [2]. Considering an isotropic elastic matrix, the linear elastic misfit is given as ҰЕ*ЎЦҰИ*/3. This energy contribution is taken into account in the present simulation. If a precipitate nucleates coherently in the bulk crystal, the entire misfit energy, as given in Eq. (3), is theoretically operative in the nucleation process. If the volumetric misfit is large enough, the stress fields can be relaxed to some extend due to matrix plasticity. For precipitates located at dislocations, a certain portion of the elastic misfit is compensated for by either the compressive or tensile component of the dislocation stress field. The effective elastic misfit used in the simulations is therefore smaller than the value for homogeneous bulk nucleation. For precipitates nucleating at grain boundaries, we assume that there is no elastic misfit involved during nucleation, since stresses are quickly relaxed by fast diffusion of vacancies and atoms along the grain boundaries [9].
The total interfacial free energy contribution (ҰӨGsurf) is equal to the specific interfacial energy (ҰГ) multiplied with the surface area of the nucleus. The interfacial energy between precipitate and matrix is a key input parameter for precipitation kinetics simulations since it has direct impact on nucleation rate and nucleation coarsening regime (Oswald ripening). As we know in solid-state precipitation, absolute values of interfacial energies cannot be measured directly through experiment, and if they can not be calculated on a physical basis, they can easily be misused and/or misinterpreted as a fitting parameter that can affect the entire precipitation behavior of the system. In the present work, the generalized nearest-neighbor broken bond model (GBB) is used to calculate the interfacial energies. It is assumed that both matrix and precipitate are homogeneous up to their common planar sharp interface. The chemical contribution to the energy of a planar sharp interface is determined by the energy of bonds across the interface and will be estimated from the enthalpy of solution of precipitates, which can be calculated from the thermodynamic database used [10]. The obtained quantity refers to the upper limit of the interfacial energy. To compensate the simplification taken as planar sharp interface, corrections considering the interfacial curvature [11], entropic effects [12] and precipitate shape effects (spheres, platelets, needles) are taken into account.
2.1 Role of vacancy evolution
Growth, coarsening and dissolution of precipitates are all diffusion-controlled mechanisms. Diffusion in aluminum alloy is governed by the vacancy-atom exchange and is directly proportional to the density of vacant lattice sites. The amount of instantaneous vacancy concentration and its time and temperature dependent evolution play a fundamental role in diffusional transport of the chemical species and therefore influence the kinetics of the whole process. Any precipitation kinetics simulation, especially in complex heat treatment or whenever natural aging is applicable, evidentially fails if detailed consideration on the vacancy effects and their evolution is ignored. The overall effect of vacancies on precipitations kinetics can be prevailing in two different topics, effect of excess vacancy evolution and effect of solute atoms vacancy traps.
2.1.1 Effect of excess vacancy evolution
If a specimen is heated to and held at some specific temperature, the population of vacancies will increase to reach the equilibrium site fraction of vacancies at that temperature. Upon cooling or heating, the vacancy annihilation and generation processes dictate whether the vacancy concentration evolves close to the equilibrium value or deviates from it. Any deviation from equilibrium values causes the instantaneous vacancy concentration to be different in comparison to equilibrium vacancy concentrations. To quantify the excess vacancy annihilation/generation at different types of sinks and sources represented by jogs at dislocations, incoherent interfaces, grain boundaries or free surfaces, a model has been developed and implemented in the thermo-kinetic software tool MatCalc [13,14].
Apart from the vacancy evolution and the effect of excess vacancies in accelerating substitutional diffusion, excess vacancies can interact with precipitation process in two other distinct mechanisms. First, if the precipitate incorporates vacancies into crystal lattice, the vacancies can be considered a chemical component which is structurally required to build up the precipitate. From this point of view, an excess of vacancies facilitates nucleation. Examples can be considered in early stage of precipitates such as vacancy-rich solute clusters and GP zones. The trapped vacancies in crystal lattice can be released again in part during dissolution of the early clusters or GP-zones. Vacancies that are released back into the still supersaturated matrix again facilitate aging, especially at low temperature [15,16]. Second, the elastic strain energy generated due to volumetric misfit of precipitates can be relaxed or partially relaxed by excess vacancy annihilation or generation at incoherent or semicoherent phase boundaries. This phenomenon in addition to flow plasticity for larger molar volume precipitates represents important stress relaxation mechanisms [17]. These stress relaxation mechanisms may lower the nucleation barrier so as to make homogeneous incoherent nucleation possible. It is noteworthy that by detail investigation of critical nucleation energy (including the chemical contribution energy of vacancies), in the case that the specific volume of the precipitate is smaller than the matrix, excess vacancies, in contrary to stress relaxing effect, effectively reduce the chemical driving force and thus act against precipitate nucleation [18].
2.1.2 Solute atoms vacancy traps
Strong energetic binding between vacancies and solute atoms lead to temporary trapping of vacancies and can directly influence diffusion in the alloy. This effect has been experimentally observed long ago by decreased response of Al-Cu to natural aging by microalloying addition of the element Sn, which has large binding energy with vacancies [19]. Industrial aluminum alloys contain, in addition to major alloying elements, minor alloying elements. If energetic binding between vacancies and these elements are high, these elements can change the free instantaneous concentration of vacancies and directly disturb diffusion in the material by solute atoms vacancy traps mechanism. Note that vacancy trapped atoms are also mobile but with different diffusivity. The overall effect is that the precipitations kinetics will be altered. This consequence that diffusion will be enhanced mostly by excess quenched-in vacancies is more pronounced for quenched and naturally aged alloys. Quantitatively accurate solute-vacancy binding energies in aluminum can be provided by first-principles calculations. The mathematical expressions for modeling have been derived by FISCHER et al [20]. The same methodology has been adapted for solute atoms vacancy trap and has been implemented recently in the software MatCalc.
2.2 Aluminum matrix
Grain size, subgrain size and dislocation density depend on the manufacturing route and the thermo-mechanical history of the material. These matrix microstructural parameters will mainly affect the number of potential nucleation sites for the semi- and incoherent precipitates. Furthermore, they act as sources and sinks for vacancies generation and annihilations.
The elastic modulus of the aluminum matrix is another important input quantity that affects the evaluation of elastic misfit stress. Its value is taken as 69.22ЎБ109-(4.01ЎБ107)T, where T is the temperature. Poisson ratio for aluminum is taken as 0.33. A uniform dislocation density of 1ЎБ1011 m-2 has been assumed for the matrix in the annealed condition.
3 Simulation set up
Specimens were taken from two types of commercial wrought alloys, 6xxx (Al-Mg-Si) and 2xxx (Al-Cu-Mg-Si). The chemical compositions of the alloys are listed in Table 1. The effects of Si pick up by Fe and Mn is taken into account by reducing the amount of Si in the kinetic simulations. Because Fe and Mn are AlFeSi phases former at rather high temperatures, we assume that these phases show no further reactions in the heat treatment. In the commercial Al-Cu-Mg-Si alloy, although the amount of Sn is very low in comparison to the other main elements (see Table 1), but due to strong energetic binding between vacancies and Sn, it has been taken into account. The effect of other trace elements has been assumed to be negligible.
Table 1 Chemical compositions of two alloys 6xxx (Al-Mg-Si) and 2xxx (Al-Cu-Mg-Si) considered for simulation

3.1 Al-Mg-Si alloy
The generic precipitation sequence that is generally accepted for Al-Mg-Si alloys is
SSSЎъ Mg and Si clustersЎъGP zones Ўъ Pre-ҰВЎдЎдЎъҰВЎдЎд ЎъҰВЎд (+BЎд+U1+U2)ЎъҰВ (4)
The early stages of precipitates that have been considered in the simulations are: early disordered metastable Mg-Si co-clusters [21], full coherent Al-containing GP-zones [22], coherent Pre-ҰВЎдЎд (Al2Mg5Si4) which is similar to ҰВЎдЎд structure but contains aluminum atoms [23], and needle-like monoclinic ҰВЎдЎд (Mg5Si6). The semi-coherent phases included in the simulation are ҰВЎд, BЎд, U1 and U2. The stable phases considered are ҰВ (Mg2Si) and Si in diamond structure [1]. In our simulation, we have considered no nucleation for ҰВЎдЎд but a transformation from Pre-ҰВЎдЎд to ҰВЎдЎд which is physically more realistic.
If we do not consider the default values existing in MatCalc, there are many different parameters that can be adjusted for each specific precipitate, concerning its nucleation, diffusion and vacancies. Always sound scientific judgment is necessary, especially for choosing those input parameters that are not fixed. For example, the applicable maximum volumetric coherency misfit for each phase can be taken from first-principle calculations but it will be reasonable if we consider some relaxation and to use the value as a fine tune up parameter for the overall better match of the simulation with experiments. As an example, for the case of ҰВЎдЎд, first-principle calculations by HUIS et al [24] indicated a lattice mismatch for different axes of 3.6%, 0.6% and 5.3 %, respectively, with an angle difference of -5.2Ўг. These results are in accordance with the results from WANG et al [25] and they corresponded to a volumetric mismatch of 9.8 %. But ҰВЎдЎд grows along the monoclinic b-axis in the direction of the lowest lattice mismatch to minimize the coherency elastic strain part of its interfacial energy. Therefore, it would be justifiable to consider an effective value of 2.5%, which satisfies the integrity of simulation, too. Or, because the morphology of a phase can change during its growth, we considered the shape factor within the limits taken from the TEM images. So to get optimized simulation we considered 10 for Pre-ҰВЎдЎд and ҰВЎдЎд and 5 for ҰВЎд and BЎд.
3.2 Al-Cu-Mg-Si alloy
Precipitation in Al-Cu-Mg-Si alloys is very diverse and the final product of the precipitation path depends strongly on chemical composition, the amount of Cu and the ratio between Mg and Si [26]. In view of the chemical composition (Table 1), we do not expect to have S (Al2CuMg) family participates. A reasonable expected precipitation sequence could be
SSSЎъClustersЎъGP zonesЎъPre-ҰВЎдЎдЎъҰВЎдЎд+ҰИЎдЎъQЎд+ҰИЎд/ҰИЎъQ+ҰИ+Si (5)
Following expected phases were considered input for our simulation: the phases for Al-Mg-Si system plus semi coherent QЎд (Al4CuMg6Si6), incoherent Q (hexagonal AlMgSiCu) and coherent ҰИЎдЎд (Al3Cu tetragonal/plate-like), semicoherent ҰИЎд (tetragonal/plate- like), incoherent stable ҰИ (tetragonal/plate-like Al2Cu) and incoherent Si. The difference between ҰИЎд and ҰИ is the coherency. Therefore, it is reasonable to assume that the phase ҰИЎд will be transformed to ҰИ and has no independent heterogeneous nucleation. To consider the effect of vacancy trap by different solute elements in the alloy, we have considered solute-vacancy binding energies as input to our simulation. Vienna ab initio simulation package (VASP) has been used for calculation of solute- vacancy binding energies and the results are listed in Table 2.
Table 2 Vacancy binding energies for elements used in Al-Cu-Mg-Si simulation

4 Experimental
Solution treatments for Al-Mg-Si and Al-Cu- Mg-Si were carried out at 540 ЎгC for 30 min and at 500 ЎгC for 60 min respectively, followed by quenching into 25 ЎгC water. The freshly quenched DSC samples were tested immediately. The DSC measurements were performed in a TA-instrument DSC-2920 in nitrogen atmosphere using a heating rate of 5 K/min for Al-Mg-Si and 10 K/min for Al-Cu-Mg-Si. DSC samples were tested vs 99.99% pure Al reference sample.
To validate the precipitation kinetics in DSC simulations, samples prepared corresponding to specific temperatures on DSC curves followed by quenching were investigated by TEM and high resolution TEM (HRTEM). TEM pictures of precipitates were taken under zone axis of <001>Al. Bright field and dark field images were taken for number counting and size measuring. The sample thickness was estimated by counting thickness fringes from the sample edge to the grain boundary.
5 Result and discussion
The result of the DSC simulation of Al-Mg-Si alloy compared to the experimental result is shown in Fig. 1(a) and the corresponding phase fraction is shown in Fig. 1(b). Although the sequence of precipitation peaks is complex, most of the simulated exothermic and endothermic peaks are in good agreement with experiment. The peaks related to GP-zones and ҰВЎдЎд simulations, which are in prime interest in industries, are almost identical with the DSC experiment. The simulation peaks occur at about the same temperature as in the experiments. In the high-temperature regime between 300 ЎгC to 400 ЎгC, there are deviations specially in fluctuation of simulation curve in comparison to experiment, which could be due to overlapping stability ranges of many different phases (ҰВЎд, U1, and U2 and BЎд), which are energetically very near to each other and the nature of numerical calculations creating the curve.

Fig. 1 Comparison between experimental and simulated DSC curves of Al-Mg-Si alloy (a) and phase fraction evolution during DSC probing of different metastable and stable phases in simulated Al-Mg-Si alloy (b)
To validate the result of the simulation, the microstructure at three specific temperatures on the experimental DSC curve, namely at 150, 248 and 303 ЎгC, which correspond to the beginning of peak II and end of peaks III and IV, respectively, are studied by TEM/HRTEM. Bright field (BF) and HRTEM results of the sample heated to 150 ЎгC did not show contrast of any precipitate, where clusters and/or GP zones are expected. This is probably due to the extreme small size of the precipitates. Precipitates at 248 ЎгC are shown in Fig. 2(a). Needle-shaped precipitates can be observed lying along <001>Al and being homogeneously distributed in the matrix. The average length of 50 randomly selected particles is 23 nm. The mean radius is 3 nm and the aspect ratio is evaluated as 7.5. The number density is quantified to be 1.9ЎБ1022 m-3.
A typical HRTEM image of a precipitate in end-on morphology at 248 ЎгC is shown in Fig. 3(a). The corresponding FFT diagram is shown in Fig. 3(b). Indexing the diffraction pattern reveals characteristics of monoclinic structure with lattice parameters a=1.51 nm, c=0.67 nm, ҰВ=105Ўг. This meets well with the reported crystal structure of ҰВЎдЎд [27]. HRTEM and FFT diagrams of other particles show only a monoclinic feature of ҰВЎдЎд, having different rotations with Al matrix. The widely reported needle-shaped ҰВЎдЎд is identified to be the only precipitate at 248 ЎгC, contributing to exothermal peak III in Fig. 1. The simulation result for  ЎдЎд shows a mean radius of 3.4 nm with a number density of 2.11ЎБ1022 m-3. Co-existence of ҰВЎд and BЎд precipitates is identified at 303 ЎгC as shown in Fig. 2(b), contributing to exothermal peak IV in Fig. 1. Both phases have similar lath shape and lay along <001> direction of the matrix and are not differentiable from their morphologies. Nevertheless, their crystal lattice parameters are different and they can be recognized in HRTEM [28].
ЎдЎд shows a mean radius of 3.4 nm with a number density of 2.11ЎБ1022 m-3. Co-existence of ҰВЎд and BЎд precipitates is identified at 303 ЎгC as shown in Fig. 2(b), contributing to exothermal peak IV in Fig. 1. Both phases have similar lath shape and lay along <001> direction of the matrix and are not differentiable from their morphologies. Nevertheless, their crystal lattice parameters are different and they can be recognized in HRTEM [28].
HRTEM results of end-on morphologies of precipitates at 303 ЎгC are shown in Fig. 4(a) and (d), respectively. Two types of precipitates are recognized, with white spots showing the same hexagonal feature but different lattice parameters. In Fig. 4(a), the spot distance along the a-axis is 0.71 nm, while 1.04 nm is found in Fig. 4(d). FFT diagram of the two images are shown in Figs. 4(b) and (e) respectively. Indexing each diffraction pattern shows crystal structure features of ҰВЎд and BЎд.
Among the 27 randomly selected precipitates in HRTEM image, 20 are identified as BЎд, leading to the conclusion that BЎд is dominant at this temperature. If we compare the phase fraction of BЎд with ҰВЎд at 303 ЎгC, we can see the same tendency (see Fig. 1(b)). We did not find any U1 or U2 phases under the studied condition. This is probably due to the low phase fraction of U1 and U2 and the close competition among U, BЎд and ҰВЎд [29,30]. Therefore, the obtained DSC precipitation simulation of the Al-Mg-Si alloy complies with the experimental DSC curve and TEM/ HRTEM observations.

Fig. 2 BF images of precipitates

Fig. 3 HRTEM results of one typical precipitate at 248 ЎгC

Fig. 4 HRTEM results of typical precipitates at 303 ЎгC
The DSC simulation of the second example (Al-Cu-Mg-Si) along with its experimental result is given in Fig. 5(a). The corresponding phase fraction of the same heat cycle is given in Fig. 5(b). There is a good agreement between the experimental DSC peaks and the simulation peaks and the positions of the simulation peaks are almost identical with the experimental DSC until the end of peak IV. In the high-temperature regime, for peak V, there is deviation and fluctuation of the simulation peak in comparison to experiment, which as explained before, could be attributed to overlapping stability ranges of many different phases (ҰИЎд, QЎд, ҰИ, Q and Si) and numerical nature of calculation method.
To check experimentally the types and evolution of the precipitations in the DSC run, five representative temperatures are chosen for TEM investigations. 120 ЎгC is almost the end of exothermic peak I; 220 ЎгC somewhere is the finish temperature of the peak II or the initial temperature of peak III; 255 ЎгC, 310 ЎгC, 410 ЎгC correspond to the end temperature of exothermic peaks III and IV and some point on the endothermic peak IV. No precipitate is observed at 120 ЎгC, neither under TEM nor HRTEM. Although our simulation shows the presence of GP zones at 120 ЎгC, due to the small size of the precipitates, no contrast is given. When the sample is heated to 220 ЎгC, needle-shaped precipitates parallel to <001>Al, indicated by arrows in Fig. 6(a) can be observed. HRTEM image at 220 ЎгC (Fig. 7(a)) show small precipitates with end-on morphology of needle (circular shapes), but also some rare in plate shape. According to our simulation we expect the beginning of the presence of QЎд and qЎд precipitations at 220 ЎгC.

Fig. 5 Comparison between experimental and simulated DSC curves of Al-Cu-Mg-Si alloy (a) and phase fraction evolution during DSC probing of different metastable and stable phases in simulated Al-Cu-Mg-Si alloy (b)
As the temperature increases from 255 ЎгC to 310 ЎгC, corresponding to Figs. 6(b) and (c), respectively, larger sizes and smaller number density of precipitates are achieved. Clear streaks from precipitates in SAED pattern can be seen only in the sample heated to 310 ЎгC. HRTEM images at 255 ЎгC (Fig. 7(b)) and 310 ЎгC (Figs. 7(c) and (d)) show the co-existence of multilayer like and circle/rectangular shaped structure, which gets thicker and the contrast of needles gets stronger. Detailed FFT studies of precipitates at 310 ЎгC under HRTEM were carried out. Both FFT studies and morphologies of the precipitates support the recognition of ҰИЎд (Fig. 7(c)) and QЎд (Fig. 7(d)) phases, respectively.
As shown in the DSC experiment (Fig. 5(a)), the overall effect of peak V is endothermic, but from the simulation (Figs. 5(a) and (b)) we see overlapping precipitation and dissolution reactions of different phases in this regime. From our TEM investigation, we see that, as the sample is heated to 410 ЎгC, all precipitates lose coherency and specific tilting is not required to make them visible in the TEM. A combination of dispersoids and different precipitates with various shapes can be observed (Fig. 6(d)). Si precipitates can be observed only at this temperature and were identified by EDX point analysis both in grains and at grain boundaries. Different precipitates with different morphologies including Al, Cu, Si bearing and Mg, Si, Cu bearing phases are recognized by EDX. Equilibrium ҰИ and Q phases expect to be among them. From the TEM and HRTEM findings at 120, 220, 255, 310 and 410 ЎгC, we can conclude to have achieved the correct sequence of precipitations in accordance with DSC simulation.

Fig. 6 BF TEM images of precipitates and corresponding SAED patterns for selected temperature samples

Fig. 7 HRTEM images of precipitate morphologies for selected temperature samples
Simulations in both alloy systems 6xxx and 2xxx give satisfactory results for most of the peaks in comparison with the experiments. There have been some iteration activities for Ў°fine tuningЎұ of the input parameters to obtain the DSC results. Ў°Fine tuningЎұ is needed due to the inherent approximations existing in the theories implemented in the simulations. Some approximations belong to the nature of CNT, some belong to TEP implemented and other approximations are included in other implemented models such as vacancy generation, annihilation and trapping.
6 Conclusions
1) The thermo-kinetic software MatCalc with the corresponding thermodynamic and diffusion databases was used for non-isothermal DSC simulation of two important heat treatable complex alloys systems of 6xxx and 2xxx. The general methodology and relevant important input parameters to be taken into account during thermo-kinetics simulations are explained in detail.
2) The evolution of the various metastable and stable phases in these systems can be well described with computer simulations, providing that the microstructural information of involved phases, characteristic of aluminum matrix and evolution of the vacancies and their interactions with solute atoms and clusters are properly taken into account.
3) During DSC simulations, the position of most peaks and the predicted heat flux evolution match well with the experimental DSC curves. We demonstrate that thermo-kinetic simulations of DSC curves can substantially aid in interpretation of DSC data, particularly in the presence of overlapping peaks.
4) Simulation of precipitation kinetics for industrial alloys is a complex task, since these materials are composed of many different alloying and trace elements. Due to the interaction of many different parameters in the precipitation kinetic simulation of industrial alloys, fine tuning of the input parameter set with sound scientific judgment is of prime importance.
5) Designing a simple suitable DSC experiment, which expresses the time-temperature dependence of the phase evolutions, provides valuable experimental information, which helps getting kinetic simulation parameters for that material. The same parameter set can be used for precipitation kinetics simulation of complex heat treatments and/or yield strength calculation of the same material.
Acknowledgment
Financial support by the Austrian Federal Government (in particular from Bundesministerium  Verkehr, Innovation und Technologie and Bundesministerium Wirtschaft, Familie und Jugend) represented by
Verkehr, Innovation und Technologie and Bundesministerium Wirtschaft, Familie und Jugend) represented by 
 mbH and the Styrian and the Tyrolean Provincial Government, represented by Steirische
mbH and the Styrian and the Tyrolean Provincial Government, represented by Steirische  mbH and Standortagentur Tirol, within the framework of the COMET Funding Programme is gratefully acknowledged.
mbH and Standortagentur Tirol, within the framework of the COMET Funding Programme is gratefully acknowledged.
References
[1] POVODEN-KARADENIZ E, LANG P, WARCZOK P, FALAHATI A, WUJUN , KOZESCHNIK E. CALPHAD modeling of metastable phases in the AlЁCMgЁCSi system [J]. Calphad, 2013, 43: 94-104.
[2] FALAHATI A, AHMADI M R, LANG P, POVODEN-KARADENIZ E, WARCZOK P, KOZESCHNIK E. Thermo-kinetic computer simulation of precipitation and age-hardening effect in Al-Mg-Si alloys with arbitrary heat treatment [C]//Materials Science and Technology (MS&T), Columbus, Ohio: MS8TЎд11, 2011: 292-299
[3] MatCalc, The Materials Calculator, 2014. http://www.matcalc.at
[4] STARINK M J. Analysis of aluminium based alloys by calorimetry: Quantitative analysis of reactions and reaction kinetics [J]. International Materials Reviews, 2004, 49(3-4): 191-226.
[5] Thermodynamic database for Al-systems, version (mc_al_v1.012.tdb) [M]. Austria: Institute of Materials Science and Technology, Vienna University of Technology, 2013.
[6] Mobility and physical databases for Al-systems, version (mc_al_v1.006.ddb and mc_al_v1.019.pdb) respectively [M]. Austria: Institute of Materials Science and Technology, Vienna University of Technology, 2012.
[7] RUSSELL K C. Nucleation in solids: The induction and steady state effects [J]. Adv Colloid Interface Sci, 1980, 13: 205-318.
[8] SVOBODA J, FISCHER F D, FRATZL P, KOZESCHNIK E. Modelling of kinetics in multi-component multi-phase systems with spherical precipitates I: Theory [J]. Mater Sci Eng A, 2004, 385: 166-174.
[9] FISCHER F D, SVOBODA J,  E, KOZESCHNIK E, SONDEREGGER B. Modelling of precipitation kinetics with simultanous stress relaxation [C]//MRS Proc. Cambridge: Cambridge University Press, 2007: 0979-HH11-04.
E, KOZESCHNIK E, SONDEREGGER B. Modelling of precipitation kinetics with simultanous stress relaxation [C]//MRS Proc. Cambridge: Cambridge University Press, 2007: 0979-HH11-04.
[10] SONDEREGGER B, KOZESCHNIK E. Generalized nearest- neighbor broken-bond analysis of randomly oriented coherent interfaces in multicomponent FCC and BCC structures [J]. Metall Mater Trans A, 2009, 40: 499-510.
[11] SONDEREGGER B, KOZESCHNIK E. Size dependence of the interfacial energy in the generalized nearest-neighbor broken-bond approach [J]. Scripta Mater, 2009, 60: 635-638.
[12] LEE Y W, AARONSON H I. Anisotropy of coherent interphase boundary energy [J]. Acta Metall, 1980, 28(4): 539-548.
[13] FISCHER F D, SVOBODA J, APPEL F, KOZESCHNIK E. Modelling of excess vacancy annihilation at different types of sinks [J]. Acta Materialia, 2011, 59(9): 3463-3472.
[14] FALAHATI A, LANG P, KOZESCHNIK E. Precipitation in Al-alloy 6016ЎӘThe role of excess vacancies [J]. Material Science Forum, 2011, 706-709: 317-322.
[15] FERRAGUT R, DUPASQUIER A, MACCHI C E, SOMOZA A, LUMLEY R N, POLMEAR I J. Vacancy-solute interactions during multiple-step ageing of an Al-Cu-Mg-Ag alloy [J]. Scr Mater, 2009, 60(3): 137-140.
[16] SANDBERG N, SLABANJA M, HOLMESTAD R. Ab initio simulations of clustering and precipitation in Al-Mg-Si alloy [J]. Computaional Materials Science, 2007, 40(3): 309-318.
[17] SVOBODA J, E, FISCHER F D. Relaxation of the elastic strain energy of misfitting inclusions due to diffusion of vacancies [J]. Philosophical Magazine Letters, 2005, 85(9): 473-479.
[18] KOZESCHNIK E. Modeling solid-state precipitation [M]. New York: Momentum Press, 2013: 72.
[19] KIMURA H, HASIGUTI R R. Interaction of vacancies with Sn atoms and the rate of G-P zone formation in an Al-Cu-Sn alloy [J]. Acta Metallurgica, 1961, 9(12): 1076-1078.
[20] FISCHER F D, SVOBODA J, KOZESCHNIK E. Interstitial diffusion in systems with multiple sorts of traps [J]. Modelling Simul Mater Sci Eng, 2013, 21(2): 1-13.
[21] STARINK M J, WANG S C. The thermodynamics of and strengthening due to co-clusters: General theory and application to the case of Al-Cu-Mg alloys [J]. Acta Mater, 2009, 57(8): 2376-2389.
[22] MATSUDA K, GAMADA H, FUJII K, UETANI Y, SATO T, KAMIO A, IKENO S. High-resolution electron microscopy on the structure of Guinier-Preston zones in an Al-1.6 mass pct Mg2Si alloy [J]. Metall Mater Trans A, 1998, 29(4): 1161-1167.
[23] HASTING H S,  A G, ANDERSEN S J, VISSERS R, WALMSLEY J C, MARIOARA C D, DANOIX F, LEFEBVRE W, HOLMESTAD R. Composition of " precipitates in Al-Mg-Si alloys by atom probe tomography and first principles calculations [J], J Appl Phys, 2009, 106(12): 123527.
A G, ANDERSEN S J, VISSERS R, WALMSLEY J C, MARIOARA C D, DANOIX F, LEFEBVRE W, HOLMESTAD R. Composition of " precipitates in Al-Mg-Si alloys by atom probe tomography and first principles calculations [J], J Appl Phys, 2009, 106(12): 123527.
[24] van HUIS M A, CHEN J H, SLUITER M H F, ZANDBERGEN H W. Phase stability and structural features of matrix-embedded hardening precipitates in Al-Mg-Si alloys in the early stages of evolution [J]. Acta Mater, 2007, 55(6): 2183-2199.
[25] WANG Y, LIU Z K, CHEN L Q, WOLVERTON C. First-principles calculations of ''-Mg5Si6/ҰБ-Al interfaces [J]. Acta Mater, 2007, 55: 5934-5947.
[26] SKIN D G. Decomposition of supersaturated solid solutions in Al-Cu-Mg-Si alloys [J]. Journal of Materials Science, 2003, 38: 279-290.
[27] MARIOARA C D, ANDERSEN S J, ZANDBERGEN H W, HOLMESTAD R. The influence of alloy composition on precipitates of the Al-Mg-Si system [J]. Metallurgical and Materials Transactions A, 2005, 36: 691-702.
[28] DUMOLT S D, LAUGHLIN D E, WILLIAMS J C. Formation of a modified beta'-phase in aluminum-alloy 6061 [J]. Scripta Met, 1984, 18(12): 1347-1350.
[29] MATSUDA K, IKENO S, MATSUI H, SATO T, TERAYAMA K, UETANI Y. Comparison of precipitates between excess SiЁCtype and balanced-type Al-Mg-Si alloys during continuous heating [J]. Metallurgial and Materials Transactions A, 2005, 36: 2007-2012.
[30] WOLVERTON C. Crystal structure and stability of complex precipitate phases in Al-Cu-Mg-(Si) and Al-Zn-Mg alloys [J]. Acta Mater, 2001, 49(16): 3129-3142.
Ahmad FALAHATI1, Jun WU1, Peter LANG2, Mohammad Reza AHMADI1, Erwin POVODEN-KARADENIZ3, Ernst KOZESCHNIK1,3
1. Institute of Materials Science and Technology, Vienna University of Technology, 9-11, 1040 Vienna, Austria;
2. Materials Center Leoben Forschung GmbH, 12, 8700 Leoben, Austria;
3. Christian Doppler Laboratory for Early Stages of Precipitation, Institute of Materials Science and Technology, Vienna University of Technology, 9-11, 1040 Vienna, Austria
ХӘ ТӘЈәАыУГІоКҫЙЁГиБҝИИ·Ё(DSC)СРҫҝІ»Н¬өД№ММ¬Паұд·ҙУҰЎЈФЪМШ¶ЁөДИИҙҰАнЦЬЖЪЦРDSCРЕәЕәНПаөДЙъіЙУлИЬҪвХэПа№ШЎЈФЪ»щУЪКұјдәНОВ¶ИИИҙҰАнЦЬЖЪЦРЈ¬DSCјмІвөҪөДКФСщөДИИСЭұдҪб№ы¶ФУЪ·ЦОцПаСЭұдәНОціц¶ҜБҰС§ј«ОӘЦШТӘЎЈАыУГИИ/¶ҜБҰС§ДЈДвФӨІвБЛ6xxxәН2xxxВБәПҪрөДИИСЭұдDSCРЕәЕЎЈҙЛДЈРНН¬КұҝјВЗБЛІ»Н¬СЗОИПаЎўОИ¶ЁПаЎўҙг»рІРУаҝХО»өДЧҙМ¬әНУ°ПмЎЈАыУГНёЙдөзҫөәНёЯ·ЦұжВКөзҫөСйЦӨDSCЗъПЯЙПМШ¶Ё·еЦөөгОціцПаөДРОіЙЎўіЯҙзәНГЬ¶ИЎЈ
№ШјьҙКЈәІоКҫЙЁГиБҝИИЈ»ВБәПҪрЈ»Оціц¶ҜБҰС§Ј»ДЈДвЈ»ҝХО»Ј»MatCalc
(Edited by Hua YANG)
Corresponding author: Ahmad FALAHATI; Tel: +43-1-58801 30812; E-mail: ahmad.falahati@tuwien.ac.at
DOI: 10.1016/S1003-6326(14)63327-6

