���±��: 1004-0609(2006)10-1778-09
fcc, hcp��bcc�ṹAu��ԭ��״̬��
�����������¶ȵı仯��ϵ
�ջԽ�, л����, ���콨, ���, �����, ������
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ��ɳ 410083)
ժ Ҫ: ��ϴ�������ԭ��(OA)���ۺ�Debye-Gr��neisenģ��, ����CALPHAD����ȷ���ľ����ȶ�����, �о���SGTE ���������ݿ���fcc, hcp��bcc �ṹAu��ԭ��״̬�� ԭ�����ܡ� ԭ�Ӷ��ܡ� ԭ������� �嵯��ģ����������ϵ���������������¶ȵı仯��ϵ�� �������: OA���۵õ���fcc-Au���ӽṹ�еĹ���d����������, �Ӷ�����fcc-Au�������ĵ����뾶�Լ�����ԭ�����; �¶�����������, 3�ֽṹ��ԭ�ӵ����뾶��С˳��Ϊ: fcc>bcc>hcp, ������Ӧ��ԭ�������С˳��Ϊ: fcc>bcc>hcp; 3�־���ṹԭ�����ܴ�С˳��Ϊ: fcc��hcp��bcc, ���һԭ��VASP������һ��; 3�ֽṹ��ԭ�Ӷ������¶ȵ����ӷ��ȴ�Լ�����ܵ�4.2��, �¶�����������ԭ�Ӷ��ܵı仯Զ�������ܡ�
�ؼ���: fcc; hcp; bcc; Au; ԭ��״̬; Debye-Gr��neisenģ��; CALPHAD����
��ͼ�����: TG111 ���ױ�ʶ��: A
Temperature dependence of atomic states and physical properties of fcc and metastable hcp and bcc Au metals
TAO Hui-jin, XIE You-qing, PENG Hong-jian, YU Fang-xin,
LIU Rui-feng, LI Xiao-bo
(School of Materials Science and Engineering,
Central South University, Changsha 410083, China)
Abstract: Combining the One-Atom(OA) theory with Debye-Gr��neisen model, the temperature dependence of atomic state, atomic potential and vibrating energy, atomic volume, bulk modulus and linear thermal expansion coefficient of fcc and metastable hcp and bcc Au metals in SGTE database of pure elements was studied with the lattice stability parameters determined by CALPHAD method. The results show that it is the smallest number of covalent electrons in d state of fcc-Au among that of the three structures that leads to the biggest single bond radii and atomic volume. The order of single bond radii of the three structures during the elevation of temperature is fcc>bcc>hcp, and it is the same order for atomic volume. The order of potential energy is fcc��hcp��bcc, and it agrees well with that by VASP program of first principles; the atomic vibrating energy obviously increases with its rate 3.2 times higher than that of potential energy when temperature increases, and the variation of atomic vibrating energy is much higher than that of atomic potential energy during temperature elevation.
Key words: fcc; hcp; bcc; Au; atomic state; Debye-Gr��neisen model; CALPHAD method
��ͼ�ļ���ģ��(CALPHAD)һֱ����Ϊ��ָ���²��Ͽ�����Ƶ�ǿ��������[1, 2], �Դ����ʲ�ͬ����ṹ���Gibbs��, �������ȶ�������������CALPHAD����Ҫ������ ����, SGTE(Scientific Group Thermodata Europe) ���������ݿ�[3]�Ѿ�����298.15K����78��Ԫ�صIJ�ͬ����ṹ��Gibbs�ܱ���ʽ�;����ȶ������� ͬʱ, �ڲ�����ʵ�����ϵ������, ͨ�����ƺ����Ͳ��������к������, ��һԭ������ȷ�������ʲ�ͬ����ṹ������ȶ��ԡ� ���о����Ѿ��õ�һԭ������ϵͳ�о���78�ִ�����fcc, hcp �� bcc�ṹ������ȶ���, ����CALPHAD�����Ľ�������˶Ա�, ������ֶ��ߴ����ű��ʵIJ��[4]��
�Ͻ�ϵͳ��ѧ���(systematic sciences of alloys, SSA)[5]�Ľ�������Ϊ�˼�С�����������ֲ��, ̽���Դ�������Ͻ�ԭ��״̬�� ����ѧ���ʺ��������ʵ�ȫ�������� ��һ����Զ�ԭ���������[6, 7]�� ����������[8]�� ԭ��״̬�ӻ�����[9, 10]�������������ʵ�����Ϊ����, ��Ag-Cu[11-14]�� Ti-Al[15-17]��Au-Cu[18-21]�ȺϽ�ϵ��ԭ��״̬, ԭ�����ܡ� ԭ�Ӷ��ܡ� ԭ������� �嵯��ģ����������ϵ�����������¶Ⱥͳɷֵı仯���ɽ������о��� ����һ�����, ������Ĺ������ڵ�ԭ��(one atom, OA)����ָ����, �Ը��ֲ�ͬ����ṹ�����ʵ�״̬���������¶ȵı仯���м���ģ�⡣ �������߲���Debye-Gr��neisenģ�ͺ�CALPHAD�����ľ����ȶ�����, ����ģ����SGTE���������ݿ���fcc, hcp��bcc-Au��ԭ��״̬�������������¶ȱ仯�Ĺ�ϵ��
1 ԭ���뷽��
1.1 ��������ԭ������
1.1.1 ԭ��״̬����
���ȶ�������ԭ�����, ����̬�����е�ԭ����ͨ����ѧ����ϵġ� ���, ����ԭ�����ĵ��ӿ����չ��ܷ�Ϊ�ɼ��ͷdzɼ����������[21]�� ��������һ�ְ������ۡ� ���ɻ���Ե��ӵĻ�ϼ�, ���ԭ�����ɼ��ļ۵��ӿ��Է�Ϊ���ۡ� ���ɺʹŵ���, �dzɼ�����(��ƷǼ�����)����Ϊ���л�ѧ���Ե�����ʵ���ӡ� ����, ���۵��ӶԽ��������Ҫ����, ���ɵ��ӶԵ��硢 ���Ⱥ���������Ҫ���ס�
��OA������, �û���ԭ��̬��k(k=1, 2, ��, n)�����ӻ����õ�������ռ����(quasi-electron-occupation, QEO)������ԭ��״̬��:

ʽ��ÿһ������̬��k����ѭPauling������ԭ��, ck���ӻ�ϵ����
�Խ���Au, sc, sf, pc, dc��dn�ֱ��ʾ��ԭ���ӻ�״̬�еĹ���s���ӡ� ����s���ӡ� ����p���ӡ� ����d���ӺͷǼ�d����; ��k��Rk(��λΪ0.1nm)�ֱ��ǹ���d�����ڳɼ������е���ռ�����Ľ��˵�Pauling�����뾶; sck, pck��dck�ֱ��ʾ��k������ԭ��̬��s, p ��d ����Ĺ��۵�����; sfkΪ���ɵ�s ̬����; dnkΪ�Ǽ���d����; nc, nf��nv�ֱ��ʾ�ܵĹ��۵��ӡ� ���ɵ��Ӻͳɼ��������� �����  ��дΪ
��дΪ , ��Au�ĵ�ԭ��״̬�������Ա�ʾΪ
, ��Au�ĵ�ԭ��״̬�������Ա�ʾΪ

1.1.2 ����������
�±�s��ȡֵ1, 2, ����ʾ����ڡ� �ν��ڡ� ���ȹ��ۼ�; Gs���ɾ���ṹ���;����ı�ʾ��ͬ����ԭ�Ӽ����뾧����������ϵ�ij���, �����fcc�ṹ, 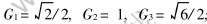 ; RΪ�����뾶; ��ΪPauling�ļ�����[22]; nc���ܵĹ��۵�����; IsΪ��s���ڼ��ĵ�ͬ����, �������̿ɱ�ʾΪ
; RΪ�����뾶; ��ΪPauling�ļ�����[22]; nc���ܵĹ��۵�����; IsΪ��s���ڼ��ĵ�ͬ����, �������̿ɱ�ʾΪ
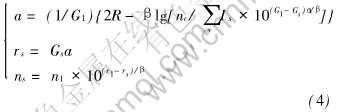
1.1.3 ����ܺ��ƺ���
��ԭ�������(Many-atom-interactions, MAI)���ܺ�����������ʽ����:
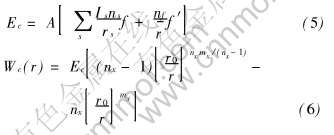
ʽ�� A�Ƿ�ӳ�����ԭ���е��ӶԺ˵������ЧӦ�ij���; 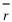 ��ʾ���ɵ��Ӽ���ƽ������, ��
��ʾ���ɵ��Ӽ���ƽ������, �� 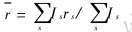 ; f��f��ֱ�������ۺ����ɵ��Ӽ��ijɼ�����, ��
; f��f��ֱ�������ۺ����ɵ��Ӽ��ijɼ�����, �� 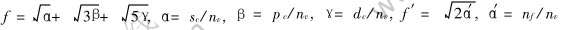 ; r0��r�ֱ����ƽ��̬������״̬����̼���; nx��mx�ֱ�ΪMAI��, ��Wx(r)�Ƶ���ϲ�����
; r0��r�ֱ����ƽ��̬������״̬����̼���; nx��mx�ֱ�ΪMAI��, ��Wx(r)�Ƶ���ϲ�����
1.1.4 �嵯��ģ������������ϵ��
�����嵯��ģ���Ķ��� 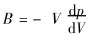 ��������ѹǿ�Ĺ�ϵ
��������ѹǿ�Ĺ�ϵ 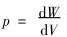 ���Եõ�
���Եõ�
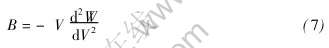
ʽ��������¶ȵı仯����Debye-Gr��neisenģ��[23]ȷ������������ϵ���ɼ���Ϊ
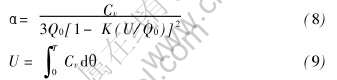
ʽ�к�������Cv�;�������U��Debye����ȷ��, Gr��neisen�����еij���K��Q0��ʵ��������ϵõ���
1.2 ����ԭ��̬
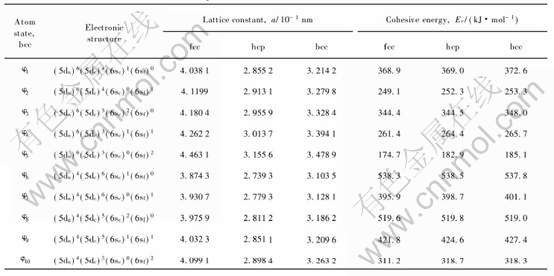
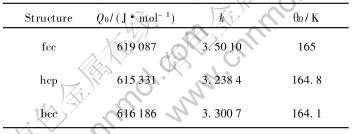
Eckardt��[24]�о�����: ���ڴ���ǿ�ҵ�s-p�ӻ�, ����Au���δռ�ݵ�p�ܼ�ʵ�����Ѿ���Ч��s�ܼ��� ���, p�ܼ��ĵ��ӿ�����Ϊs̬���ӡ� �������ֽ��Ʒ���, ���ļ�����10�ֻ���ԭ��̬����Ӧ�IJ�ͬ����ṹ�ľ������ͽ����, ����hcp�ṹ�����c/a��Ϊ�����1.633, ���������ڱ�1��
2 ������
2.1 ԭ��״̬
ͼ1�������Ծ������ͽ����Ϊ��, ���ӻ����������ҵ���a�ߺ͵�Ec�ߵĽ���, �����ݽ�������̬�ӻ��ɷ�, ���ȷ��ԭ��״̬�Ĺ���, ��2�������ӻ��ɷּ���Ӧ���ʡ�
���������ӻ��ɷֿ���ȷ��fcc, hcp��bcc-Au�ĵ��ӽṹ�͵����뾶�ֱ�Ϊ

��1 ����Au�Ļ���ԭ��̬�Ͳ�ͬ����ṹ�ľ������ͽ����
Table 1 Lattice constants and cohesive energies of fcc, hcp and bcc-Au in basic atomic state
��2 fcc, hcp��bcc-Au��̬�ӻ��ɷֺ�����
Table 2 Composition and properties of fcc, hcp and bcc-Au

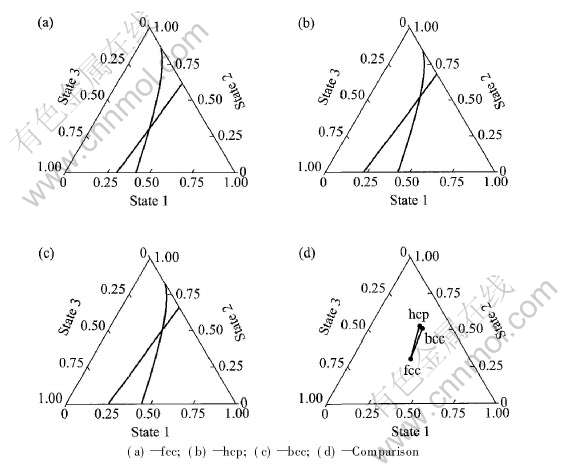
ͼ1 fcc, hcp��bcc-Au��̬�ӻ��ɷ�ͼ
Fig.1 Composition positions of three state hybridization of fcc, hcp and bcc-Au
2.2 ��������
2.2.1 ���ܺ���
nx��mx����MAI�ƺ���Wx(r)�е�ָ��, r0Ϊƽ��״̬�����ԭ�Ӽ���, EcΪ�����, ��fcc, hcp��bcc-Au��Wx(r)�����3��ͼ2��ʾ��
2.2.2 ��������ϵ�����嵯��ģ��
Debye-Gr��neisenģ�Ͳ������4��ʾ, ��������ϵ���е�ʵ�����ݲμ�����[23], ͼ3������ ��������ϵ���� �������� ԭ������� �����뾶���嵯��ģ�����¶ȵı仯���ߡ�
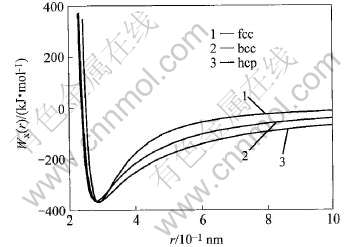
ͼ2 fcc, hcp��bcc-Au����������
Fig.2 Potential energy curves of fcc, hcp and bcc-Au
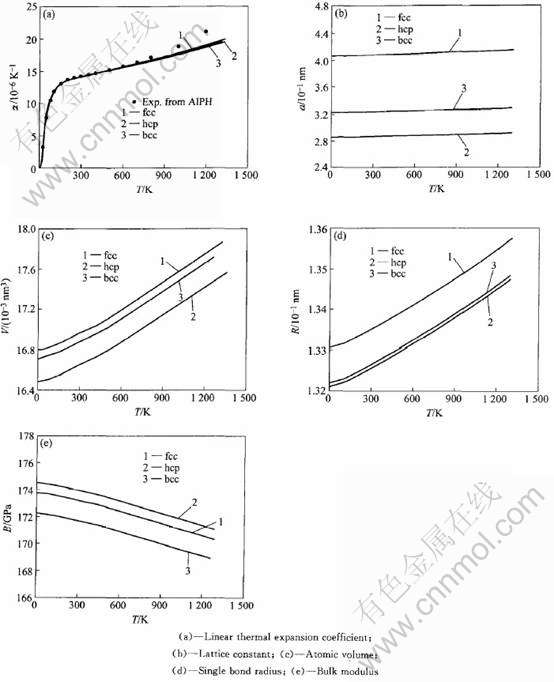
ͼ3 fcc, hcp��bcc-Au����������ϵ������������ ԭ������� �����뾶���嵯��ģ�����¶ȵı仯
Fig.3 Temperature dependence of linear thermal expansion coefficient,
lattice constant, atomic volume, single bond radii and bulk modulus of fcc, hcp and bcc-Au
��3 fcc, hcp��bcc-Au��Wx(r)�ƺ���������
Table 3 Parameters of potential function of fcc, hcp and bcc-Au

��4 fcc, hcp��bcc-Au Debye-Gr��neisenģ���о��ļ������
Table 4 Constants of linear thermal expansion for fcc, hcp and bcc pure Au metal
2.2.3 ԭ�Ӷ��ܺ�����
ԭ�Ӷ��ܺ��������¶ȵı仯��ϵ��ͼ4��
Ϊ�˶��������ʽ��ж����Ƚ�, ��ͼ3��4��ȡ0K, 298.15K���۵�����ݽ��жԱ�, ������ڱ�5��
3 ����������
3.1 ԭ��״̬
���Ľ�OA���۷���������һԭ��CAS-
TEP(Cambridge sequential total energy package)������������Ԫ����(LRC)[24]�Ľ�������˶Ա�(��6), �������: OA����hcp��bcc-Au�Ľ�����һԭ���dz��ӽ�, fcc-Au����d���������ƫ��, ����Ҫԭ����fcc-Au���ӽṹ�еĹ���d������ƫ�͡� ��һ�������fcc-Au���������ֽṹ�ĵ����뾶��Ҫ��, �Ӷ�ԭ��������
�����¶�����, ���ӽṹ�����ȶ�, �����뾶���¶����������͡� ����������, 3�ֽṹ��ԭ�ӵ����뾶��С˳��ʼ�ձ���Ϊ: fcc>bcc>hcp�� ԭ�������Ϊ���ۺ�ƽ�������ϵĸ���, �ɾ���ṹ�͵����뾶��ͬ����, ���Ҹ��ݱ��Ľ��֪������Au��ԭ�������Ҫ�ɵ����뾶����, �����������ԭ�������Сͬ��Ϊ: fcc>bcc>hcp��
3.2 ��������
Ϊ�˶����Ͻ����һ������, �б�Ҫ���ɵ��ӽṹ�������������������������Ľ�����жԱȡ� ���Ľ�0 KʱOA���۵Ľ��(�������ļ�
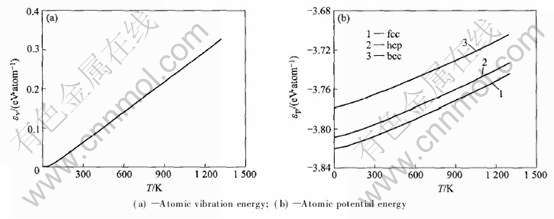
ͼ4 fcc, hcp��bcc-Au��ԭ�Ӷ��ܺ��������¶ȵı仯
Fig.4 Temperature dependence of atomic vibration energy and potential energy of fcc, hcp and bcc-Au
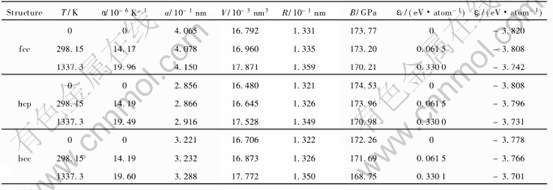
��5 fcc, hcp��bcc-Au��0K, 298.15K���۵���������ʶԱ�
Table 5 Properties of fcc, hcp and bcc-Au at 0K, 298.15K and 1337.33K
��6 ��ͬ������������fcc, hcp��bcc-Au��0Kԭ��״̬
Table 6 Comparison of atomic states of fcc, hcp and bcc-Au at 0K by various methods

��7 ��ͬ�����о�����fcc, hcp��bcc-Au 0Kʱ�������ʵĶԱ�
Table 7 Calculated properties of fcc, hcp and bcc-Au at 0K by different methods
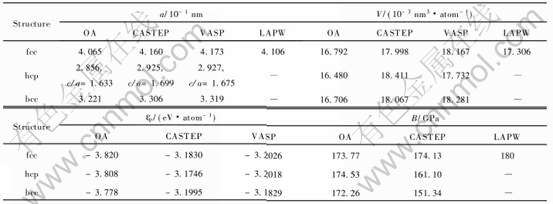
�㷽��������[29])���һԭ��CASTEP��VASP����[4]�Լ�������ƽ�沨LAPW����[27]�Ľ�������˶Ա�, ������ڱ�7��
�����϶Աȿ�֪: OA�õ���fcc�ṹ�ľ�������ԭ�������LAPW�����ӽ�, CASTEP��VASP�����ӽ�; OA�õ���3�ֽṹԭ�������С˳��Ϊ: fcc>bcc>hcp, ��VASP�����ӽ�, ��hcp�ṹԭ�������С, ����fcc>hcp, ����Ҫ����Ϊԭ�ӵ����뾶�IJ�����ɵ�; ����ԭ������, OA�����VASPһ��, ���СΪ: fcc��hcp��bcc, �������ȶ���Ϊ: fcc>hcp>bcc; �����嵯��ģ��, OA��CASTEP���������bcc�ṹֵ��С�� �ڵ��ӽṹ�ȽϽӽ��������, bcc��hcp�ṹ�����ܲ�����п����dzɼ�����Ũ�Ȳ�����ɵ�, Ϊ��, ���干��d����Ũ��Tcd=ncd/V�� ����s����Ũ��Tcs=ncs/V�����ɵ���Ũ��Tf=nf/V��3���оݽ��жԱ�, ������ڱ�8�� �ɱ�8��֪: ���ֽṹ�����ɵ���Ũ��Tf����С, ͬʱ, hcp��С��ԭ����������乲�۵���Ũ��, �ر���Tcdֵ��bcc�Ĵ�, �Ӷ�����ܸ���, ������ȶ��������¶�����, �嵯��ģ��B��С, ���С˳��Ϊ: hcp>fcc>bcc, ����Ҫ����3�ֽṹDebye�¶Ⱥ�ԭ��������������µġ� ����Debye�¶ȵIJ���, ������Debye-Gr��neisenģ�ͼ���ʱ, 3�ֽṹ����������ϵ����ԭ�Ӷ�������С�� ��ͼ4�ͱ�5��֪: 3�ֽṹ��ԭ�Ӷ������¶ȵ����ӷ�����ԭ�����ܵ�4.2����, �����¶���������ԭ�Ӷ��ܵı仯Զ�������ܡ�
��8 hcp��bcc-Au�ɼ�����Ũ�ȵĶԱ�
Table 8 Concentration difference of bonding electrons in hcp and bcc-Au(nm-3)

4 ����
1) �ںϽ�ϵͳ��ѧ(SSA)�����, ���ô�������ԭ��(OA)���ۺ�Debye-Gr��neisenģ�Ͷ�fcc, hcp��bcc-Au�ĵ��ӽṹ�������о�, ����OA���۵õ���hcp��bcc-Au�Ľ�����һԭ���õ��ķdz��ӽ�, ��fcc-Au��d���������ƫ��, �����ԭ����fcc-Au���ӽṹ�еĹ���d������ƫ��, ���fcc-Au�������ĵ����뾶, �Լ�����ԭ�������
2) ԭ�������Ϊ���ۺ�ƽ�������ϵĸ���, �ɾ���ṹ�͵����뾶��ͬ����, ���Ľ������������Au��ԭ�������Ҫ�ɵ����뾶����, ���¶�����������, 3�ֽṹ��ԭ�ӵ����뾶��С˳��Ϊ: fcc>bcc>hcp, ��Ӧ��ԭ�������СΪ: fcc>bcc>hcp��
3) ͨ�����һԭ��������жԱ�, ����OA���ۼ����3�־���ṹԭ�����ܵĽ�����һԭ��VASP����һ��, ��ԭ�����ܴ�СΪfcc��hcp��bcc, �Ӷ�����3�ֽṹ�ľ����ȶ���Ϊ: fcc>hcp>bcc; hcp��bcc�ṹ������˳����ԭ�����˳���෴����Ҫԭ������2�ֽṹ�Ĺ��۵���Ũ�ȴ��ڽϴ���졣
4) 3�ֽṹ��ԭ�Ӷ������¶ȵ����ӷ��ȴ�Լ�����ܵ�4.2��, �¶�����������ԭ�Ӷ��ܵı仯Զ��������; ��������ϵ����ԭ�Ӷ��ܲ���С����Ҫԭ������3�ֽṹ��Debye�¶ȷdz��ӽ���
REFERENCES
[1]Kaufman L, Bernstein H. Computer Calculation of Phase Diagram[M]. New York: Academic Press Inc, 1970.
[2]Saunders N, Miodownik A P. CALPHAD (CalAulation of PhaseDiagrams): A Comprehensive Guide[M]. New York: Pergamon, Oxford, 1998.
[3]Dinsdale A T. SGTE data for pure elements[J].CALPHAD, 1991, 15(4): 317-425.
[4]Wang Y, Aurtarolo S, Jiang C, et al. Ab initio lattice stability[J] in comparison with CALPHAD lattice stability. CALPHAD, 2004, 28: 79-90.
[5]XIE Y Q, TAO H J, PENG H J, et al. Atomic states, potential energies, volumes, stability and brittleness of ordered fcc TiAl2 type alloys[J]. Physica B, 2005, 366: 17-37.
[6]XIE Y Q. A new potential function with Many-Atom interactions in solid[J]. Science in China(series E), 1993, 36(1): 90-99.
[7]XIE Y Q. Relationship of Lennard-Jones potential and Morse potential with Wx(r) potential[J]. Trans Nonferrous Met Soc China, 1994, 4(4): 63-66.
[8]XIE Y Q, ZHANG X D, ZHAO LY, et al. Electronic structure and properties of Au metal[J]. Science in China(series A), 1993, 36(4): 487-494.
[9]XIE Y Q, MA L Y, ZHANG X D, et al. Microstructure and properties of Au-Ni alloys[J]. Science in China(series A), 1993, 36(5): 612-623.
[10]XIE Y Q. Electronic structure and properties of pure iron[J]. Acta Metallurgica Materialia, 1994, 42(11): 3705-3715.
[11]XIE Y Q. Atomic energies and Gibbs energy functions for Ag-Au alloys[J]. Science in China(series E), 1998, 41(2): 146-156.
[12]XIE Y Q, ZHANG X D. Atomic volumes and volume functions for Ag-Au alloys[J]. Science in China(series E), 1998, 41(2): 157-168.
[13]XIE Y Q, ZHANG X D. Electronic structure of Ag-Au alloys[J]. Science in China(series E), 1998, 41(3): 225-236.
[14]XIE Y Q, ZHANG X D. Phase diagram and thermodynamic properties of Ag-Au alloys[J]. Science in China(series E), 1998, 41(4): 348-356.
[15]XIE Y Q, PENG K, LIU X B. Influences of xTi/xAl on atomic states, lattice constants and potential-energy planes of ordered fcc TiAl-type alloys[J]. Physica B, 2004, 344: 5-20.
[16]XIE Y Q, LIU X B, PENG K, et al. Atomic states, potential energies, volumes, stability, and brittleness of ordered fcc TiAl3-type alloys[J]. Physica B, 2004, 353: 15-33.
[17]XIE Y Q, PENG H J, LIU X B, et al. Atomic states, potential energies, volumes, stability and brittleness of ordered fcc Ti3Al-type alloys[J]. Physica B, 2004, 362: 1-17.
[18]YU Fang-xin, XIE You-qing, NIE Yao-zhuang. Electronic structure of Au-Au alloys[J]. Trans Nonferrous Met Soc China, 2004, 14(6): 1041-1049.
[19]л����. Au-Cu�Ͻ�ϵ�������������ľ�����[J]. ����ѧ��, 1998, 34(12): 1233-1242.
XIE You-qing. Lattice constants of disordered and ordered phases in the Au-Cu system[J]. Acta Metallurgica Sinica, 1998, 34(12): 1233-1242.
[20]л����, ������. Au-Cu�Ͻ���۽ṹ������[J]. ����ѧ��, 1994, 30(12): 531-539.
XIE You-qing, ZHANG Xiao-dong. Microstructure and properties of Au-Cu alloys[J]. Acta Metallurgica Sinica, 1994, 30(12): 531-539.
[21]GUO Y Q, YU R H, ZHANG R L, et al. Calculation of magnetic properties and analysis of valence electronic structures of LaTi3-xAlx(T=Fe, Co) compounds[J]. Journal of Physical Chemistry B, 1998, 102(1): 9-16.
[22]Pauling L. The Nature of the Chemical Bond[M]. Ithaca: Cornell University Press, 1960.
[23]Kirby R K, Hahn T A, Rothroch B D. Thermal expansion[A]. Gray D E. American Institute of Physics Handbook[C]. USA: McGraw-Hill Book Company, 1972: 4-119-4-138.
[24]Eckardt H, Fritsche L, Noffke J. Self-consistent relativistic band structure of the noble metals[J]. J Phys F: Met Phys, 1984, 14: 97-112.
[25]Kittel C. Solid State Physics[M]. New York: John Wiley and Sons Inc, 1976.
[26]Ozolin V, Wolverton S C, Zunger A. Au-Au, Ag-Au, Au-Ag, and Ni-Au intermetallics: First-principles study of temperature-composition phase diagrams and structures[J]. Phys Rev B, 1998, 57(11): 6427-6443.
[27]Wei S H, Mbaye A A, Ferreira L G, et al. First-principles calculations of the diagrams of noble metals: Au-Au, Au-Ag and Ag-Au[J]. Phys Rev B, 1987, 36(8): 4163-4185.
[28]�¾���, ��л�. ������Ͻ��еĹ�̬���[M]. ����: ұ��ҵ������, 1997: 8-10.
Chen Jing-rong, LI Cheng-ji. Phase Transitions of Solids in Metals and Alloys[M]. Beijing: Metallurgical Industry Press, 1997: 8-10.
[29]XIE You-qing, DENG Yong-ping, LIU Xin-bi. Electronic structure and physical properties of Cr, Mo, W metal[J]. Trans Nonferrous Met Soc China, 2003, 13(5): 1102- 1107.
(�༭������)
������Ŀ: ������Ȼ��ѧ����������Ŀ(50271085; 50471058)
�ո�����: 2006-02-13; ������: 2006-04-30
ͨѶ����: л����, ����; �绰: 0731-8879287; E-mail: xieyouq@mail.csu.edu.cn

