文章编号:1004-0609(2013)04-1160-08
氧、氢和碳原子在α-铀(001)表面吸附与扩散特性的
第一性原理研究
刘智骁,邓辉球,胡望宇
(湖南大学 物理与微电子科学学院,长沙 410082)
摘 要:应用第一性原理密度泛函理论系统研究氧、氢和碳原子在α-铀(001)表面的吸附与扩散特性。研究发现:在铀表面氧原子与氢原子择优吸附在H2位置,碳原子倾向于占据在H1位置;氧原子在铀表面的扩散势垒较低,容易在铀表面上扩散,形成表面氧化层;氢原子的扩散势垒较高,碳原子的扩散势垒最大,难以在表面扩散。吸附原子从铀表面向次表面层扩散时,氧原子的扩散势垒很高,难以向次表面扩散;碳和氢原子的扩散势垒较低,特别是在氧的辅助作用下,碳原子向次表面的扩散势垒降低约0.5 eV,使碳原子易于向次表面层扩散;铀表面上会形成氧化层,次表面会形成富碳层,可对铀的进一步氧化起到抑制作用,这与相关实验结果符合较好。
关键词:α-铀;第一性原理;表面吸附;扩散势垒
中图分类号:O485 文献标志码:A
First-principles study of adsorption and diffusion properties of
O, H and C atoms on α-U(001) surface
LIU Zhi-xiao, DENG Hui-qiu, HU Wang-yu
(School of Physics and Microelectronics, Hunan University, Changsha 410082, China)
Abstract: The adsorption and diffusion properties of O, H and C atoms on α-U(001) surface were studied by first-principles density functional theory approach. For the on-surface adsorption, O and H atoms prefer to occupy the H2 site, while C atoms tend to occupy the H1 site. The diffusion barrier of O atom on U surface is low and the diffusion is easy, which will lead to the formation of uranium-oxidation layer on the top of surface. The diffusion barrier of H atom is larger and that of C atom is the largest one, thus it is difficult for C atom diffusion on the U surface. As for the diffusion of atom from the surface site to the sub-surface, the barriers of H and C atoms are low, while that of O atom is very high, which indicates that the O atom can not migrate into the next surface. And the barrier of C atom diffusion to the sub-surface is reduced by about 0.5 eV with the assistance of O atom nearby. The U-O oxidation layer on the surface and the U-C layer on the sub-surface can be formed easily, which help to prevent the metallic U from further oxidation. These results agree good with available experiments.
Key words: α-uranium; first-principles; surface adsorption; diffusion barrier
基金项目:国家自然科学基金资助项目(10976009)
收稿日期:2012-06-06;修订日期:2012-12-28
通信作者:邓辉球,教授,博士;电话:0731-88823226;E-mail: hqdeng@hnu.edu.cn
铀(U)是在自然界中存在的最重的化学元素,原子序数为92,价电子排布为5f36d17s2。未排满的5f壳层会与6d7s壳层发生杂化,使得铀具有奇异的几何结构和电子结构,并能以复杂的方式成键[1]。单质铀存在3种结构[2]:常温常压下,基态铀为底心正交的α相;在940~1 050 K的温度区间内,晶体铀转变为复杂的底心正交(BCT)的β相;温度在 1 050 K直至熔点,铀以体心立方(BCC)的γ相存在。铀的化学性质非常活泼,极易在空气中发生氧化还原反应,导致表面被腐蚀;铀还能和许多金属反应生成金属间化合物[3]。铀一旦在表面发生氧化腐蚀反应,则其化学性质和核反应能力都要受到影响,极大地影响了金属铀的贮存和安全使用。
气体分子在铀表面的吸附特性一直是实验的研究热点。MCLEAN等[4]研究了O2 在多晶α-U表面的吸附行为,发现氧原子会停留在铀表面上,并形成铀的氧化物。HASCHKE[5]在前人实验工作的基础上总结了环境湿度与温度对铀腐蚀速率的影响,他指出铀在干燥氧气中的腐蚀速率最慢,在水蒸汽中的腐蚀速率最快,而在潮湿氧气中的腐蚀速率介于二者之间。实验认为常温下CO和CO2 在金属铀表面也是化学解离吸附, 即CO和CO2 气体首先在清洁的金属铀表面解离吸附,表层形成氧化物;解离的碳原子通过扩散进入金属铀基体内并在亚表层形成铀碳化物(UC),表面则形成过氧化物(UO2+x)[4, 6]。
近年来,随着计算机技术的发展,大规模并行计算与密度泛函理论相结合,使得人们可以从电子尺度上研究气体分子与金属表面的相互作用。近年来,较多的研究者采用密度泛函理论研究气体分子与金 属[7-13]及其表面[14-15]的相互作用,但是研究含氧气体在铀表面吸附的工作却较少。RAY等[16-17]研究了O2 与CO在γ-U(001)表面的吸附特性,发现CO以C做吸附端的分子吸附结构最稳定,而O2 则会在γ-U(001)表面发生解离吸附。李赣等[18-20]研究了CO、O2 和CO2在α-U(001)表面的吸附特性,指出气体分子的解离吸附在热力学上更稳定,且氧分子在铀表面的解离不存在解离势垒。NIE等[21]应用第一性原理计算了氢分子在α-U(001)表面的吸附与解离特性,发现氢分子的解离势垒小于0.34 eV。
综上所述,大气环境中铀表面吸附的气体分子极易解离为原子,因此,研究氧、氢和碳原子在铀表面的吸附扩散特性对了解铀表面的性质有重要的意义。本文作者采用第一性原理密度泛函理论方法系统研究了氧、氢和碳原子在α-U(001)表面的择优占位以及其在近表面层间的扩散性质,该工作对研究气体分子在金属铀及其氧化物表面的吸附解离过程和反应动力学特性,了解金属铀表面与各种气体的氧化腐蚀反应机理,并寻找能使金属铀表面钝化或缓解其被腐蚀的气体环境的有效方法,具有一定的指导作用。
1 计算方法
在本工作中,所有的计算都采用了第一性原理密度泛函理论[22-23]结合平面波基组的方法[24-25]。求解Korn-Sham方程时使用了Vienna Ab-initio Simulation Package (VASP)软件包[26-27],并用缀加平面波法[28-29]描述电子与原子核之间的相互作用,用广义梯度近似下的PBE泛函[30]描述电子与电子之间的交换关联作用。布里渊区的K点取样采取了Monkhorst-Pack[31]方法。结构优化计算使用了共轭梯度算法[32],当原子间相互作用力不大于0.02 eV/?时,认为体系达到了力的收敛判据标准。在处理波函数的占据数时,采用了二阶Methfessel-Paxton (MP)方法[33]。在所有的计算中,平面波基组的截断能为380 eV,K点网格密度为7×7×1。由于铀原子5f电子的强关联性质,处理UO2体系时需要采用DFT+U的方法才能准确预测其基态性质[34-36]。实验结果表明,在金属铀材料中,f-f电子的关联效应并不像其铀氧化物(UO2)那样明显[37]。忽略自旋轨道耦合与电子的强关联作用,Adak用简单的DFT方法计算了高压条件下金属铀的性质,其理论预测与实验结果符合很好[38]。本文作者的测试结果也表明,对纯金属铀和原子在铀表面的吸附,可忽略5f电子关联性的影响。
在进行吸附计算时,本文作者建立了含有5层铀原子的C(2×1)表面单胞平板模型,并弛豫表面两层原子,固定下面3层原子。收敛性测试证明在两个平板之间添加12?真空层时足以用于模拟原子在铀表面的吸附。在C(2×1)表面上,考虑了吸附原子的5种典型初始吸附位置(见图1(a)):(A) 吸附原子位于最表层3个铀原子的中心并有两个次表面配位铀原子(H1);(B) 吸附原子位于最表层3个铀原子的中心并且只有一个次表面配位铀原子(H2);(C) 吸附原子位于两个次近邻铀原子的中点(LB);(D) 吸附原子位于两个最近邻铀原子的中点(SB);(E) 吸附原子位于一个铀原子的顶端(TOP)。由于吸附原子只放在铀原子松弛的一侧,会诱发出一个偶极矩,因此,在计算中本文作者加入1个偶极修正以消除因表面结构不对称而产生的偶极矩[39]。吸附能定义为
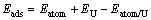 (1)
(1)
式中:Eatom为在12.0 ?×12.1 ?×12.2 ?盒子中孤立原子(氧、氢、碳)的总能;EU为清洁铀平板模型的总能;而Eatom/U为吸附后整个体系的总能;Eads即为吸附过程中释放出的能量,其数值越大,表明吸附越容易发生。
在本工作中,还研究了氧、氢和碳原子占据在表面下八面体间隙位(见图1(b))的形成能与扩散特性。这些间隙原子的形成能计算方法与吸附能的计算方法相同:
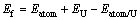 (2)
(2)
在搜寻原子扩散的最小能量路径(MEP)时,使用了CI-NEB[40]方法。计算过程中,在初态与末态间线性插入9个插值点,插值点间的弹性常数为5.0 eV/?2。
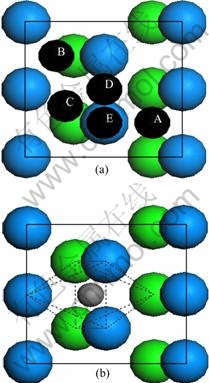
图1 C(2×1)表面的典型初始吸附位置
Fig. 1 Typical adsorption sites of C(2×1) surface: (a) On- surface adsorption site; (b) Near-surface interstice
2 结果与讨论
2.1 氧、氢和碳原子在铀表面的吸附与扩散
表1~3所列分别为氧、氢和碳原子在C(2×1)表面经优化后的吸附能与几何参数。其中,IS为吸附原子的初始吸附位置,FS为吸附原子优化后的最终吸附位置,Eads为吸附能(eV),d为吸附原子与其最近邻铀原子之间的距离(?)。对于氧原子位于H1位或TOP位的初始吸附构型,经优化后氧原子都会择优占据在H1位,这时对应的吸附能为8.06 eV,氧与其最近邻铀原子之间的距离(d)为2.21 ?。当氧原子初始置于LB位置时,经优化后的最终吸附位置也为H1位置,然而此时的吸附能略大,为8.10 eV。吸附能增大的原因是氧与铀的结合作用更强,在几何结构上表现为氧铀键长缩短0.02 ? (d = 2.14 ?)。当氧原子初始位于H2或SB位时,经优化后氧原子都会占据H2位置。对于氧原子在α-铀(001)表面的吸附,氧原子最终占据H2位时的吸附能最大,为8.15 eV,氧铀键长为2.16 ?。李赣等[9]关于氧气在α-铀(001)吸附解离的计算结果表明,解离后的氧原子都占据H2位置时吸附能更大。在本工作中,氧铀键长为2.14 ?到2.21 ?,与氧气解离吸附后氧铀键长的计算结果一致(2.12 ?到 2.45 ?)。文献[20]中还报道了单个氧原子在C(2×1)表面的吸附特性,同样发现氧原子在H2位置的吸附能比在H1位置的吸附能大0.12 eV,与本研究的结果一致(0.09 eV)。
同氧原子在α-铀(001)表面吸附一样,氢原子在该表面吸附时的最优吸附位置也是H2位。在该吸附构型中,氢原子的吸附能约为2.84 eV,它与最近邻铀原子原子间的氢铀键长约为2.24 ?。当氢原子初始置于H1位或者TOP位时,优化后氢原子都吸附在H1位,这是氢在α-铀(001)表面吸附的次稳定位置,吸附能为2.78 eV,比最优吸附位置的吸附能小0.06 eV,然而该构型与最优吸附构型的氢铀键长没有差别。应用PW91泛函,NIE等[21]计算了氢原子在只含3层铀原子的α-铀(001)表面平板模型的吸附特性。其计算结果表明,无论是在C(2×1)还是在C(1×1)表面单胞上吸附,H2位都为最优吸附位置,且H1和H2两种吸附构型的氢铀键长几乎一致(差别小于0.02 ?)。可见,聂锦兰等采用更为简单的计算模型所得的结果与本工作中氢原子在铀表面以上吸附的结果一致。
表1 氧原子在铀表面吸附优化后的吸附能与几何参数
Table 1 Optimized adsorption energies and geometric parameters of adsorbed O atom on uranium surface

IS stands for initial adsorption sites of adsorbate atoms, FS stands for finally optimized adsorption sites; Eads is adsorption energy, and d is distance between adsorbate and its nearest U atom.
表2 氢原子在铀表面吸附优化后的吸附能与几何参数
Table 2 Optimized adsorption energies and geometric parameters of adsorbed H atom on uranium surface

表3 碳原子在铀表面吸附优化后的吸附能与几何参数
Table 3 Optimized adsorption energies and geometric parameters of adsorbed C atom on uranium surface

对于碳原子在α-铀(001)表面的吸附,其最优吸附位置为H1位置,与氧、氢在铀表面的最优吸附位置不同。碳原子经优化后位于H1位置时的吸附能最大,为7.26 eV。此时碳与最近邻铀原子之间的键长为2.18 ?。当碳原子被置于H1、SB和TOP位时,优化后碳原子都会停留在H2位。H2位是碳在铀表面吸附的次稳定位置,对应的吸附能为7.18 eV,碳铀键长为2.15 ?。JIN等[10]计算了碳原子在C(2×1)的吸附特性,同样发现碳在H1位的吸附能比在H2位的吸附能大0.09 eV,且碳铀键长约为2.17 ?。
从吸附能的角度来看,氧原子与铀表面的相互作用最强,与碳的次之,氢与铀表面的相互作用最弱。通过对不同吸附原子最优吸附构型的几何结构分析,也能得到类似的规律:氧或者碳与铀的相互作用很强,因此吸附原子与铀原子的键长较短(约2.15 ?),而氢原子与铀原子的相互作用较弱,因此氢铀键较长(约2.25 ?)。
图2列出了3种原子在铀表面最优吸附构型的态密度分布(LDOS)。当氧原子吸附在α-铀(001)表面时,会使铀表面的电子发生明显的重新分布。U 5f 和6d能带会发生劈裂,在-5.5~-4.5 eV的区域产生两个吸附峰。两个吸附峰与O 2p轨道重合,表明铀和吸附的氧之间存在着明显的2p-5f-6d杂化。这种杂化普遍存在于UO2晶体[41]以及氧原子在钚表面的吸附结构 中[42-44]。当氢原子吸附在α-铀(001)表面时,铀受吸附作用而产生的电子重新分布较和氧吸附相比不是很明显。氢原子的1s轨道分布在-5~-4 eV能级之间,并与铀原子的6d轨道重合。由此可见,氢原子在铀表面发生吸附后是以1s-6d的杂化方式形成化学键。与氧在铀表面吸附的LDOS相比,U 5f电子并没有参与杂化成键,且氢铀键的6d电子成分要明显小于氧铀键中的6d电子成分,可见氧原子与铀原子的相互作用要明显强于氢原子原子铀的相互作用。RAY等[45-47]用全电子势的方法计算了氢气在dhpc-镅(0001)面、α-钚(020)面和δ-钚(111)的吸附特性,其电子结构分析表明氢气吸附在dhpc-镅(0001)面时没有1s-6d-5f杂化发生,氢气在钚表面吸附时只能引发Pu 5f6d微弱的重新分布,故氢与锕系元素的相互作用是十分微弱的。碳原子在α-U(001)表面发生吸附以后,C 2p轨道与U 5f6p轨道完全重合,意味着碳原子和铀原子之间以2p-5f-6d的杂化方式成键。此外,与氧或者氢吸附相比,碳原子吸附并没有在费米能级下-5 eV左右诱发出U 5f6d电子的重新分布,然而,在-8 eV处,出现了一个很小的6d电子分布区域。李赣等[20]分析了碳原子与氧原子在α-铀(001)表面共吸附的LDOS,发现C 2p轨道只与费米能级附近的U 5f6d能带有重合,而与O 2p轨道及其激发的新的U 5f6d电子分布区域(费米能级以下4.4 eV到5.6 eV)没有重合。RAY等[48-49]也指出碳原子在δ-钚(111)和(100)表面吸附时,只在费米能级附近发生2p-5f-6d杂化。从LDOS分布可以看出,吸附原子可以引发铀电子结构的变化,其中氧的作用最明显,氢的作用最弱。这种规律可能是由3种原子的电负性不同决定的(氧、碳、氢原子的电负性分别为3.44、2.55、2.20)。
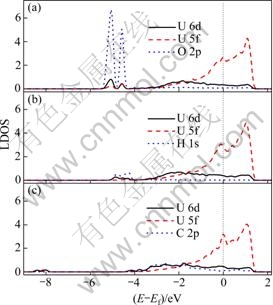
图2 3种原子在C(2×1)表面最优吸附位置局域态密度分布
Fig. 2 DOS of favorite configurations of adsorbates on C(2×1) surface: (a) O adsorbed on H2 site; (b) H adsorbed on H2 site; (c) C adsorbed on H1 site
使用CI-NEB方法,本文作者计算了氧、氢、碳原子在α-铀(001)表面最稳定吸附位置向次稳定吸附位置扩散所要跨越的势垒(见图3)。氧原子从次稳定H1位扩散到最稳定的H2位只需要跨越0.09 eV的势垒;氢原子从次稳定的H1位扩散到最稳定的H2位所要跨越的势垒较高,为0.20 eV;碳原子从次稳定的H2位扩散到最稳定的H1位所需要的跨越的势垒最大,为0.61 eV。李赣等[19]计算得到的氧原子从H1位扩散到H2位的势垒为0.14 eV,碳原子从H2位扩散到H1位的势垒为0.57 eV,NIE等[21]计算得到的氢原子在铀表的扩散势垒约为0.18 eV。文献中报道的扩散势垒与本工作中的理论计算值相差小于0.05 eV。通过对比3种原子扩散势垒的高低,发现氧原子在铀表面最容易发生扩散,氢原子次之,碳原子相对难以在铀表面发生扩散。尤其是氢、氧两种原子的扩散势垒不大于0.20 eV,在室温条件下氢和氧原子能在铀表面迅速扩散。氧原子在铀表面的快速扩散,极易在金属铀表面形成氧化层,降低金属铀的化学和物理特性。
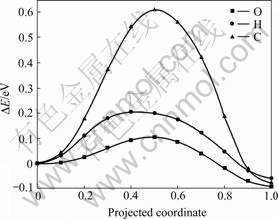
图3 O、H、C在铀表面的扩散势垒
Fig. 3 Diffusion barriers of O, H and C on uranium surface
2.2 氧、氢和碳原子在近表面间隙位的占位与扩散
在本工作中,考虑了氧、氢和碳原子在近表面间隙位的占位以及扩散特性。表4中列出了3种原子占据近表面间隙位的形成能。当氧原子占据次表面铀原子之间的间隙位(IN1)时,形成能为6.51 eV。氢原子和碳原子占据IN1为的形成能分别为1.97 eV和7.25 eV。NIE等[21]计算了氢原子位于次表面铀原子间隙位时的形成能:对于C(2×1)表面单胞模型,形成能约为2.17 eV,对于C(1×1)表面单胞模型,形成能约为2.03 eV。与吸附在表面上相比,氧原子占据IN1时的形成能下降了1.64 eV,氢原子占据IN1时的形成能下降了0.88 eV,而碳原子占据IN1时的形成能只下降了0.01 eV。分别对比氧、氢和碳原子在IN1位的形成能,可发现在热力学角度上碳比氧更容易占据近表面间隙位。图4所示为3种原子分别占据在第一二层铀原子围出的八面体间隙位的几何结构示意图。其中,编号为A、B、C的铀原子位于最外层,编号为D、E、F的铀原子为次外层原子。当氧原子占据IN1位时,其与A原子的键长为2.53 ?,与B、C原子的键长均为2.23 ?,与D原子的键长为2.61 ?,与E、F原子的键长均为2.29 ?;当氢原子占据IN 1位时,其与A原子的键长为2.06 ?, 与B、C原子的键长均为2.15 ?,与D原子的键长为3.39 ?,与E、F原子的键长均为2.22 ?;当碳原子占据IN1位时,其与A原子的键长为2.48 ?,与B、C原子的键长均为2.25 ?,与D原子的键长为2.55 ?,与E、F原子的键长为2.29 ?。总体来看,杂质原子与第一层铀原子的距离要小于其与第二层铀原子的距离。
表4 O、H、C原子在近表面间隙位的形成能
Table 4 Formation energies of O, H and C occupying near-surface interstice

应用CI-NEB方法,研究了氧、氢和碳原子由表面向近表面间隙位扩散的动力学特性(见图5)。氧原子从H1位扩散到IN1需要跨越1.97 eV的势垒,而氢原子和碳原子从H1扩散到IN1只需要跨越0.95 eV的能垒,说明碳原子和氢原子更容易从表面向近表面间隙位扩散。NIE等[21]计算得到的氢原子从H2位扩散到近表面间隙位的势垒约为0.95 eV,与本工作中以H1位为起点得到的扩散势垒相同。综上所述可见,氧原子很难扩散到次表层,因此,会在金属铀表面形成富氧层。
实验发现,铀表面上会形成富氧的铀氧化物 层,而在近表面会形成富碳的铀碳层[50],其结构如图4所示。从前面的计算可知,碳原子从表面向次表层的扩散势垒依然较高(0.95 eV),因此,本文作者研究了碳原子附近有氧原子存在时碳向次表层的扩散势垒。图6所示为氧辅助作用下碳原子向次表层的扩散势垒。由图6可知,当氧原子存在时,碳原子向次表层扩散的势垒只有0.49 eV,比无氧原子存在时降低
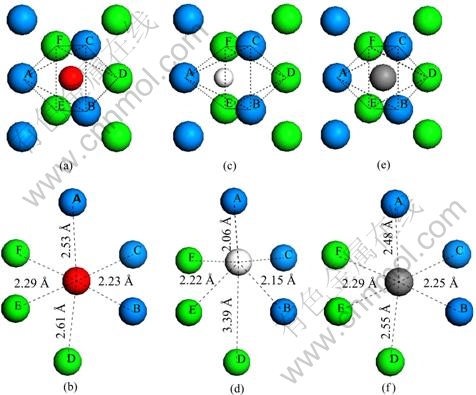
图4 氧、氢和碳原子占据IN 1间隙位的几何结构
Fig. 4 Geometric structures of O, H and C atoms occupying IN1 interstitial sites: (a), (b) O atom occupying IN 1 site; (c), (d) H atom occupying IN1 site; (e), (f) C atom occupying IN1 site
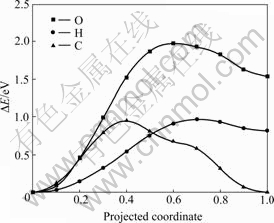
图5 氧、氢和碳原子向铀近表面间隙位扩散的势垒
Fig. 5 Diffusion barriers of O, H and C atoms from on-surface site to near-surface interstice
了约0.5 eV。结果表明,氧原子的存在降低碳原子向次表层的扩散势垒,在次表层形成富碳层。致密的富碳的碳铀化合物组断层可以阻止铀表面的进一步氧化,对保护金属铀的性质起到一定作用,与实验结果一致[50]。
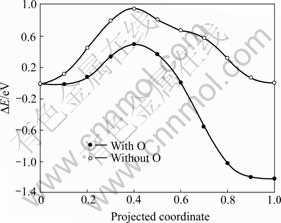
图6 氧辅助作用下碳原子向次表层的扩散势垒
Fig. 6 Diffusion barriers of C atom from on-surface site to near-surface interstice with and without O assistance
3 结论
1) 应用第一性原理计算的方法系统的研究了氧、氢、碳原子在α-铀(001)表面以及近表面的吸附与扩散特性。对于表面上吸附,氧原子与氢原子择优占据在H2位置,碳原子择优占位在H1位置。氧原子和铀原子之间以O 2p-U 5f6d形式杂化成键,氧原子与铀表面的结合能力最强。
2) 应用CI-NEB方法,得到了氧、氢和碳原子在α-铀(001)表面上的扩散势垒,发现氢原子在铀表面的扩散势垒较低,但向次表层的扩散势垒较大,难以向内扩散;氧原子在铀表面的扩散势垒非常低(0.09 eV),而向次表层扩散的势垒则很高(1.95 eV),所以氧原子非常容易在铀表面发生扩散并形成铀氧化物层;碳原子在铀表面和次表面的扩散势垒都较高,但在周围氧原子的辅助作用下,碳原子向次表层扩散的势垒大幅降低,容易从表面扩散到次表层,形成富碳的碳铀化合物阻断层,对金属铀的进一步氧化起到一定的抑制作用。
REFERENCES
[1] S?DERLIND P, ERIKSSON O, JOHANSSON B, WILLS J M, BORNING A M. A unified picture of the crystal structures of metals[J]. Nature, 1995, 374: 524-525.
[2] YOUNG D A. Phase diagrams of the elements[M]. Berkley: University of California Press, 1991.
[3] 李冠兴, 武 胜. 核燃料[M]. 北京: 化学工业出版社, 2007.
LI Guan-xing, WU Sheng. Nuclear fuel [M]. Beijing: Chemical Industry Press, 2007.
[4] MCLEAN W, COLMENARES C A, SMITH R L. Electron-spectroscopy studies of clean thorium and uranium surfaces: Chemisorption and initial stages of reaction with O2, CO and CO2 [J]. Physical Review B, 1982, 25: 8-24.
[5] HASCHKE J M. Corrosion of uranium in air and water vapor: Consequences for environmental dispersal [J]. Journal of Alloys Compound, 1998, 278: 149-160.
[6] 伏晓国, 汪小琳, 刘柯钊. 金属铀与CO原位反应的X射线光电子能谱研究[J]. 稀有金属, 2002, 26: 249-252.
FU Xiao-guo, WANG Xiao-lin, LIU Ke-zhao. In-situ study of surface reaction of uranium with carbon monoxide[J]. Chinese Journal of Rare Metals, 2002, 26: 249-252.
[7] ZHANG L, ZHANG Y, GENG W T, LU G H. Towards theoretical connection between tensile strength of a grain boundary and segregated impurity concentration: Helium in iron as an example[J]. Europhysics Letters, 2012, 98: 17001.
[8] ZHOU H B, JIN S, LIU Y L, LU G H, Liu F. Stress tensor: A quantitative indicator of effective volume and stability of helium in metals[J]. Europhysics Letters, 2011, 96: 66001.
[9] ALKHAMEES A, LIU Y L, ZHOU H B, JIN S, ZHANG Y, LU G H. First-principles investigation on dissolution and diffusion of oxygen in tungsten[J]. Journal of Nuclear Materials, 2009, 393: 508-512.
[10] JIN S, LIU Y L, ZHOU H B, ZHANG Y, LU G H. First-principles investigation on the effect of carbon on hydrogen trapping in tungsten[J]. Journal of Nuclear Materials, 2011, 415: S709-712.
[11] LIU Y L, ZHANG Y, ZHOU H B, LU G H. Vacancy trapping mechanism for hydrogen bubble formation in metal[J]. Physical Review B, 2009, 79: 172103.
[12] ZHOU H B, LIU Y L, JIN S, ZHANG Y, LUO G N, LU G H. Towards suppressing H blistering by investigating the physical origin of the H-He interaction in W[J]. Nuclear Fusion, 2010, 50: 115010.
[13] ZHOU H B, LIU Y L, JIN S, ZHANG Y, LUO G N, LU G H. Investigating behaviors of H in a W grain boundary by first-principles: from dissolution and diffusion to a trapping mechanism[J]. Nuclear Fusion, 2010, 50: 025016.
[14] MA X F, SU H Y, DENG H Q, LI W X. Carbon monoxide adsorption and dissociation on Mn-decorated Rh(111) and Rh(553) surfaces: A first-principles study[J]. Catalysis Today, 2011, 160: 228-233.
[15] MA X F, DEMG H Q, YANG M M, LI W X. Atomic and molecular adsorption on RhMn alloy surface: A first principles study[J]. Journal of Chemical Physics, 2008, 129: 244711.
[16] DHOLABHAI P P, RAY A K. A density functional study of carbon monoxide adsorption on (100) surface of γ-uranium[J]. Journal of Alloys Compound, 2007, 444/445: 356-362.
[17] HUDA M N, RAY A K. Density functional study of O2 adsorption on (100) surface of γ-uranium[J]. International Journal of Quantum Chemistry, 2005, 102: 98-105.
[18] 李 赣, 罗文华, 陈虎翅. O2在α-U(001)面吸附的密度泛函理论研究[J]. 化学应用与研究, 2010, 22(10): 1283-1289.
LI Gan, LUO Wen-hua, CHEN Hu-chi. Density functional study of O2 adsorption on α-U(001)surface[J]. Chemical Research and Application, 2010, 22(10): 1283-1289.
[19] 李 赣, 罗文华, 陈虎翅. CO在α-U(001)表面的吸附[J]. 物理化学学报, 2010, 26(5): 1378-1384.
LI Gan, LUO Wen-hua, CHEN Hu-chi. CO Adsorption on α-U(001) surface[J]. Acta Physico-Chimica Sinica, 2010, 26(5): 1378-1384.
[20] 李 赣, 罗文华, 陈虎翅. CO2在α-U(001)表面的吸附和解离[J]. 物理化学学报, 2011, 27(10): 2319-2325.
LI Gan, LUO Wen-hua, CHEN Hu-chi. Adsorption and dissociation of CO2 on the α-U(001) surface[J]. Acta Physico-Chimica Sinica, 2011, 27(10): 2319-2325.
[21] NIE J L, XIAO H Y, ZU X T, GAO F. Hydrogen adsorption, dissociation and diffusion on the α-U(001) surface[J]. Journal of Physics: Condensed Matter, 2008, 20: 455001.
[22] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review A, 1965, 140(4): 1133-1138.
[23] JONES R O. The density functional formalism, its applications and prospects[J]. Review of Modern Physics, 1989, 61: 689-746.
[24] CAR R, PARRINELLO M. Unified approach for Molecular dynamics and density-Functional theory[J]. Physical Review Letters, 1985, 55: 2471-2474.
[25] PAYNE M C, TELER M P, ARIAS T A, JOANNOPOULOS J D. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients[J]. Review of Modern Physics, 1992, 64: 1045-1097
[26] KRESSE G, HAFNER J. Ab initio molecular dynamics for open-shell transition metals[J]. Physical Review B, 1993, 48: 13115-13118.
[27] KRESSE G, FURTHMLLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54: 11169-11186.
[28] KRESSE G, JOUBERt D. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Physical Review B, 1999, 59: 1758-1775.
[29] BL?CHL P E. Projector augmented-wave method[J]. Physical Review B, 1994, 50: 17953-17979.
[30] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77: 3865-3868.
[31] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13: 5188-5192.
[32] PRESS W H, FLANNERY B P, TEUKOLSKY S A, VETTERLING W T. Numerical recipes[M]. New York: Cambridge University Press, 1986.
[33] METHFESSEL M, PAXTON A. T. High-precision sampling for Brillouin-zone integration in metals[J]. Physical Review B, 1989, 40: 3616-3621.
[34] DUDAREV S L, MANH D N, SUTTON A P. Effect of Mott-Hubbard correlations on the electronic structure and structural stability of uranium dioxide[J]. Philosophical Magazine B, 1997, 75: 613-628.
[35] DUDAREV S L, BOTTON G A, SAVRASOV S Y, HUMPHREYS C J, SUTTON A P. Electron-energy-loss spectra and the structural stability of nickel oxide:?An LSDA+U study[J]. Physical Review B, 1998, 57: 1505-1508.
[36] DUDAREV S L, BOTTON G A, SAVRASOV S Y, SZOTEK Z, TEMMERMAN W M, SUTTON A P. Electronic structure and elastic properties of strongly correlated metal oxides from first principles: LSDA+U, SIC-LSDA and EELS study of UO2 and NiO[J]. Physical Status Solidi A, 1998, 166: 429-443.
[37] LANDER G H. FISHER E S, BADER S D. The solid-state properties of uranium: A historical perspective and review[J]. Advances in Physics 1994, 43: 1-111.
[38] ADAK S, NAKOTTE H, de CHAPTEL P F, KIEFER B. Uranium at high pressure from first principles[J]. Physica B, 2011, 406: 3342-3347.
[39] NEUGEBAUER J, SHEFFLER M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K layers on Al(111)[J]. Physical Review B, 1992. 46: 16067-16080.
[40] HENKELMAN G, UBERUAGA B P, J?NSSON H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths[J]. Journal of Chemical Physics, 2000, 113: 9901-9904
[41] CHEN Q Y, LAI X C, TANG T, CHU M F, ZHANG Y B, TAN S Y. First-principles study of the electronic structure and optical properties of UO2[J]. Journal of Nuclear Materials, 2010, 401: 118-123.
[42] ATTA-FYNN R, RAY A K. Relaxation of the (111) surface of δ-Pu and effects on atomic adsorption: An ab initio study[J]. Physica B, 2007, 400: 307-316.
[43] HUDA M N, RAY A K. A density functional study of molecular oxygen adsorption and reaction barrier on Pu (100) surface[J]. Eur Phys J B, 2005, 43: 131-142
[44] ATTA-FYNN R, RAY A K. Atomic adsorption on the (020) surface of α-Pu: A density functional study[J]. Physical Review B, 2005, 77: 085105.
[45] DHOLABHAI P P, RAY A K. Adsorption and dissociation of molecular hydrogen on the (0001) surface of double hexagonal close packed americium[J]. The European Physical Journal B, 2009, 67: 183-192.
[46] ISLAM M F, RAY A K. An ab initio study of H adsorption on the (020) surface of α-Pu[J]. Physical Status Solidi B, 2011, 248(1): 193-202.
[47] HUDA M N, RAY A K. Molecular hydrogen adsorption and dissociation on the plutonium (111) surface[J]. Physical Review B, 2005, 72: 085101.
[48] ATTA-FYNN R, RAY A K. A full-potential linearized augmented plane wave (FP-LAPW) study of atomic carbon, nitrogen, and oxygen chemisorptions on the (100) surface of δ-Pu[J]. Physica B, 2007, 392: 112-126.
[49] ATTA-FYNN R, RAY A K. Ab initio full-potential fully relativistic study of atomic carbon, nitrogen, and oxygen chemisorption on the (111) surface of δ-Pu[J]. Physical Review B, 2007, 75: 195112.
[50] 杨江荣, 蒋春丽, 陆 雷, 肖 红, 汪小琳, 因子分析技术在铀与CO,O2反应俄歇分析中的运用[J]. 核化学与放射化学, 2005, 27: 198-202.
YANG Jiang-rong, JIANG Chun-li, LU Lei, XIAO Hong, WANG Xiao-lin. Application of factor analysis to AES studies of uranium oxidation by CO and O2[J]. Journal of Nuclear and Radiochemistry, 2005, 27: 198-202.
(编辑 龙怀中)

