J. Cent. South Univ. (2016) 23: 286-292
DOI: 10.1007/s11771-016-3072-6
Thermodynamic analysis and dynamics simulation on reaction of Al2O and AlCl2 with carbon under vacuum
LU Yong(¬��)1, 2, ZHOU Yue-zhen(������)1, 2, CHEN Xiu-min(������)1, 2, LI Zi-yong(������)1, 2,
YU Qing-chun(���ഺ)1, 2, LIU Da-chun(����)1, 2, YANG Bin(���)1, 2, XU Bao-qiang(�챦ǿ)1, 2
1. National Engineering Laboratory for Vacuum Metallurgy
(Kunming University of Science and Technology), Kunming 650093, China;
2. Yunnan Provincial Key Laboratory of Nonferrous Vacuum Metallurgy, Kunming 650093, China
 Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Abstract: The feasibility study of the AlCl(g) generated by Al2O-AlCl2-C system under vacuum was carried out by thermodynamic analysis and CASTEP package of the Material Studio program which was based on density functional theory (DFT) formalism. Thermodynamic calculations indicate that AlCl and CO molecules can be formed under conditions of temperature 1760 K and the pressure of 60 Pa. The interaction of Al2O and AlCl2 with C shows that the chemical adsorption of Al2O and AlCl2 does take place on C(001) crystal plane, and at the same time, new chemical bond is formed between Al atom in Al2O and Cl atoms from one of the Al��Cl bonds in AlCl2. The results, after 1.25 ps dynamics simulation, indicate that adsorbed AlCl molecules are generated and CO molecule will be formed in this system, and they will escape from C(001) surface after a longer period of dynamic simulation time. It means that the reaction of Al2O and AlCl2 with C can be carried out under given constraint condition.
Key words: Ab initio molecular dynamics; carbothermic-chlorination (AlCl2) reaction; thermodynamics; interaction; Al2O
1 Introduction
Currently, the Hall-H��roult process by dissolving Al2O3 in fused NaF-AlF3 (cryolite) followed by direct current electrolysis is still the only commercial process for the production of aluminum. The high energy consumption with the aluminum production [1-2] has led numerous attempts to find alternative routes for aluminum production. The direct carbothermal reduction has the potential to reduce energy consumption by up to 38%, capital costs by more than 60%, and decrease CO2 emissions by up to 30% [3]. Meanwhile, it also reduces overall operating costs by 25%-30%, and has no perfluorocarbons (PFCs) [4]. In spite of considerable effort, the carbothermic reduction of Al2O3 to Al remains to be a formidable technical challenge, because of the high temperature required and aluminum carbide and oxycarbide byproducts formed [5].
KRUESI et al [6] and VISHNEVETSKY et al [7] pointed out that volatile Al2O can be formed in solar aluminum production process under vacuum. Recently, Kunming University of Science and Technology with support from Joint Funds of the National Natural Science Foundation of China has been researching and developing the technology to produce aluminum by indirect carbothermic reduction simultaneous chlorination of alumina under vacuum condition, in which AlCl generated by carbothermal reduction- chlorination can decompose to liquid Al and AlCl3 gas which can be easily to be separated at lower temperature zone in vacuum furnace. It can be shown by the following reactions [8-11]:

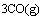 (1)
(1)
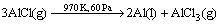 (2)
(2)
FENG et al [9] proposed that carbothermal reduction-chlorination of alumina was a successive step, in which alumina firstly reacted with carbon to generate gaseous Al2O, Al, CO in the crucible; Secondly, gaseous AlCl would be formed by chlorination (AlCl3 used as chlorinating agent) of Al2O, Al gases. It can be further deduced that Al2O, Al but not Al2O3 participate in the carbothermic-chlorination process.However, the mechanism of the reaction for Al2O(g) with intermediate AlCl2 molecule in the presence of carbon, especially under vacuum atmosphere, has not been studied up to now. The present work is aimed at investigating the thermodynamics and kinetics of the formation of AlCl according to the following reaction(3) by vacuum and to further enrich basic theory for the process of aluminum production by carbothermal- chlorination reduction of alumina.
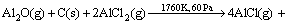
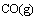 (3)
(3)
2 Calculation method and model
2.1 Thermodynamic calculation
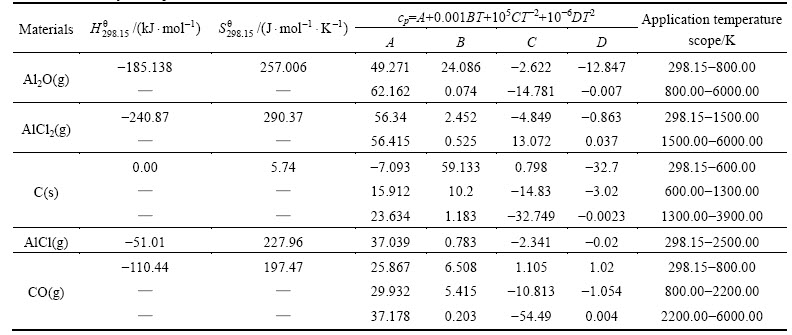
The relationships between standard state Gibbs free energy change estimated in the temperature range of 800.00-2500.00 K per 100 K (the manual setting value) and temperature for the reaction (3) under ambient pressure (100 kPa) were calculated by HSC chemistry 6.0 software with ��Gibbs free energy function method�� [12] shown from (4) to (8). The thermodynamic parameters used in this work are given in table 1 [13-15].
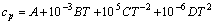 (4)
(4)
where A, B, C and D are coefficients estimated from experimental data
 (5)
(5)
where HT is enthalpy of formation at temperature T; Hf (298.15 K) is enthalpy of formation at 298.15 K; Htr is enthalpy of transformation.
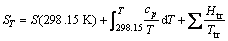 (6)
(6)
where ST is entropy of substance at temperature T; S(298.15 K) is standard entropy of substance; Ttr is temperature of phase change.
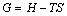 (7)
(7)
where G is Gibbs free energy.
standard Gibbs energy of reaction is
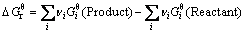 (8)
(8)
where 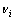 and
and 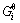 is stoichiometric number and standard Gibbs free energy of related substance i, respectively.
is stoichiometric number and standard Gibbs free energy of related substance i, respectively.
Based on the atmospheric pressure calculation, the relationships of the non-standard Gibbs free energy change vs temperature under the pressure of 10, 102, 103 and 104 Pa have been calculated by
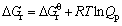 (9)
(9)
where Qp is the reaction quotient. For the reaction(3) if it is in the vacuum situation, and the residual pressure is supposed to equal the sum of the partial pressure of Al2O, AlCl2, AlCl and that of CO is P, pressure of the gaseous components is
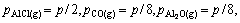
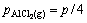
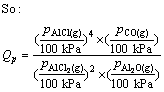 (10)
(10)
2.2 Theoretical calculation
In this work, theoretical calculations have been performed by using CASTEP package of the Material
Studio program which is based on density functional theory (DFT) formalism, the electron wave function was used by plane-wave basis set, and the energy cutoff was 300 eV [16]. The electron�Celectron interaction was treated with generalized gradient approximation (GGA) by Perdew, Burke and Ernzerhof (PBE) [17-19] exchange-correlation potential. The ultrasoft pseudopotential method was used [20]. The convergence criterion was as follows: the energy change below 2��10-5 eV/atom and displacement change less than 0.0002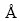 . The NPT ensemble, incliding temperature 1760 K, pressure 60 Pa, time step 1 fs, the simulation time 1 ps, the Andersen pressure controlling method [21-22] and Nos�� temperature control method [23], was used by ab initio molecular dynamic(AIMD) method [24]. The reaction (3) model structure was optimized by Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm, then the obtained stable configuration was used to calculate the energy and ab initio molecular dynamics simulation, and all of the calculations were completed in the medium grid scatter.
. The NPT ensemble, incliding temperature 1760 K, pressure 60 Pa, time step 1 fs, the simulation time 1 ps, the Andersen pressure controlling method [21-22] and Nos�� temperature control method [23], was used by ab initio molecular dynamic(AIMD) method [24]. The reaction (3) model structure was optimized by Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm, then the obtained stable configuration was used to calculate the energy and ab initio molecular dynamics simulation, and all of the calculations were completed in the medium grid scatter.
Table 1 Thermodynamic parameters used in this work
The primitive cell model of C was imported from structure database provided by Material Studio program itself. With the primitive cell, the C(001) crystal plane was cleaved, and the thickness of the C(001) crystal plane was adjusted to different conductions. On this basis, the supercell was built, and the vacuum layer set was 15.0. Then, the Al2O molecule and AlCl2 molecule were added to the supercell as the initial structure to calculate the stable structure and the properties.
3 Results and discussion
3.1 Results of thermodynamic calculation
The relationships between Gibbs free energy change (��G) and temperature for the reaction (3) were calculated at atmospheric pressure and different vacuum degrees as shown in Fig. 1. As can be seen, ��G value of reaction (3) decreased with increasing the temperature under a certain pressure and with decreasing the system pressure at a constant temperature in the range of 800.00-2500.00 K. Thereby, the higher the temperature was, and the lower the system pressure was, the more easily the forward reaction was performed. Additionally, according to Le Chatelier��s principle, vacuum was beneficial for reaction (3) because it was a volume expansion process. The spontaneous reaction temperatures Tstart at which Gibbs free energy change for reaction (3) equaled zero calculated via the theoretic formula (11) were 920.22, 1014.81, 1131.08, 1277.44, 1467.31 K under the pressures of 101, 102, 103, 104 and 105 Pa, respectively. Therefore, it was concluded that the reaction for Al2O(g) with intermediate AlCl2 in the presence of carbon could be carried out under conditions of temperature 1760 K and the pressure of 60 Pa.
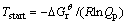 (11)
(11)
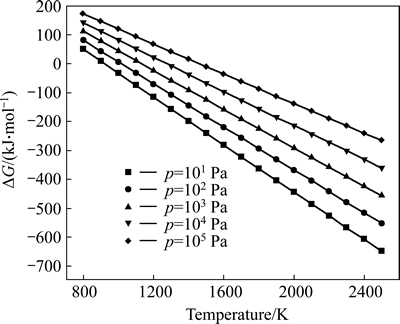
Fig. 1 Relations between ��G and T for reaction (3) at different pressures
3.2 interaction of Al2O and AlCl2 with C
Figure 2 represents the optimized stable structure of free Al2O molecule, AlCl2 molecule, and the adsorbed state of Al2O and AlCl2 on C(001) crystal plane.
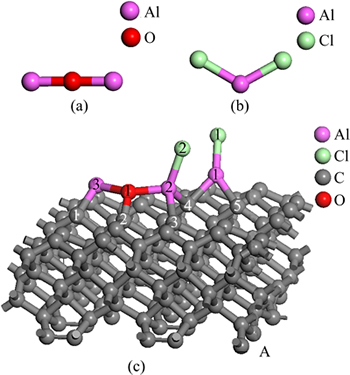
Fig. 2 Optimized stable structures of Al2O (a), AlCl2 (b) and Al2O with AlCl2 on C(001) (c)
The Mulliken populations and bond lengths of the free Al2O, AlCl2, and Al2O with AlCl2 on C(001) are given in tables 2-4.
Using the figures shown in table 4, we could know that the Mulliken populations between Al atoms and C atoms, and bond lengths were in the range 0.41-0.71, 1.92209-2.06810, separately. At the same time, the population and bond length of O and C were 0.50, 1.50174, respectively. Combined with the stable structure of Fig. 2(a) and Fig. 2(c), it could be concluded that new chemical bond had been formed among Al, O and C, namely, the chemical adsorption of Al2O molecule and AlCl2 molecule did take place on C(001) crystal plane.
Table 2 Mulliken populations and bond lengths of free state Al2O molecule
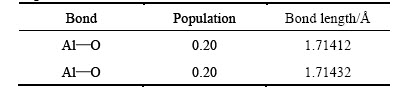
Table 3 Mulliken populations and bond lengths of free state AlCl2 molecule
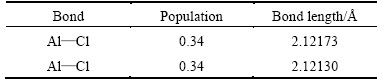
Table 4 Mulliken populations and bond lengths of Al2O with AlCl2 on C(001)
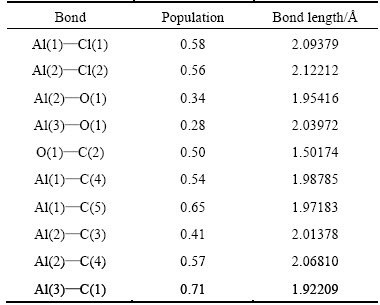
By comparing table 4 and table 2, it could be found that the Mulliken populations and bond lengths between Al and O both slightly increased, which was mainly caused by the bond between Cl(2) and Al(2). It could be considered that Al2O molecule still existed in the molecular form instead of dissociation form by comparing Fig. 2(c) with Fig. 2(a). The bond length of Al(1)��Cl(2) measured was 3.15292, obviously longer than Al��Cl bonds length in table 3. Combining the comparison of the structure between Fig. 2(c) and Fig. 2(b), Al(1)��Cl(2) bond could be determined to be broken and then a new Al��Cl bond between Al(2) atom and Cl(2) atom had formed. This meant that adsorbed AlCl2 molecule on substrate C(001) surface had been dissociated in the presence of Al2O molecule.
Figure 3 represents charge density difference of the free state Al2O after optimization, in which Fig. 3(a) is stereogram and Fig. 3(b) is sectional drawing. It could be intuitively found that electrons mainly gathered between O atom and Al atoms and clustered around O atom from Fig. 3. Thereby, the bonds between O and Al were ionic bonds, with a small amount of covalent component.
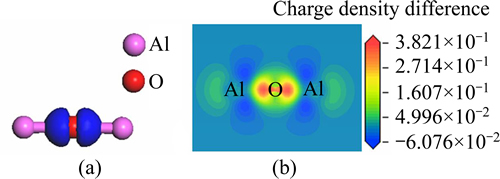
Fig. 3 Charge density difference map of free state Al2O optimized:
Figure 4 shows charge density difference of the optimized free state AlCl2, in which Fig. 4(a) is stereogram and Fig. 4(b) is sectional drawing. According to Fig. 4, we could obviously see that electrons primarily distributed between Cl atoms and Al atom and were partial to Cl atoms. Thus, the bonds between Cl and Al were covalent bonds.
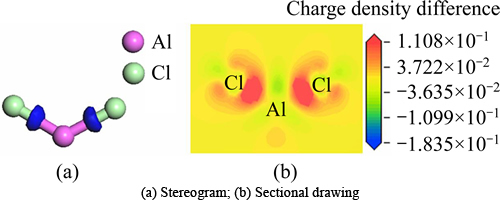
Fig. 4 Charge density difference map of free state AlCl2 optimized:
Figure 5 presents charge density difference of optimized stable structure of Al2O molecule and AlCl2 molecule on C(001) surface. In terms of O(1) and Al(2), Al(3), although charge density difference between Al and O is slightly different from that of Fig. 3, the characteristics of bonds was similar to that of free Al2O. To Al(1) and Cl(1), Al(2) and Cl(2), the type of bonds was analogous to that of Al-Cl bonds in free state AlCl2. We could intuitively found that electrons primarily distributed between O(1) and C(2) and were partial to O(1) atom. Thus, the bond between C and O was polarized covalent bond. Concerning Al(1) and C(4), C(5); Al(2) and C(3), C(4); Al(3) and C(1), as can be seen from Fig. 5, electrons mainly converged between C and Al, and clustered around C atoms. Therefore, the bonds between C and Al were ionic bonds, with a small amount of covalent component. According to the above, new chemical bond had been formed between Al and C, that is to say, the chemical adsorption existed on C(001) crystal plane. This analytical result agreed with that of the Mulliken populations.
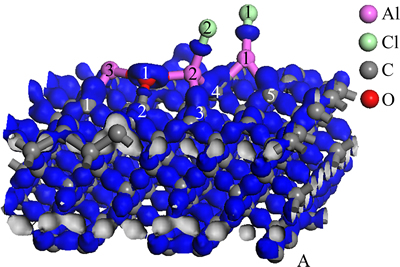
Fig. 5 Charge density difference map of Al2O and AlCl2 on C(001) surface after being optimized
3.3 Dynamic simulation of Al2O and AlCl2 with C
Figure 6 represents the structure of AlCl2 molecule and Al2O molecule on C(001) crystal plane after 1 ps dynamics simulation. The model of AlCl2 and Al2O on C(001) surface changed obviously by comparing the structure in Fig. 2(c). The bond length of O(1)��Al(3) measured was 5.25124, obviously longer than the Al��O bond lengths in table 2. Therefore, O(1)��Al(3) bond could be considered to be in dissociation. Double bond formed between O(1) atom and C(2) atom was analogous to the characteristics of the free CO molecule and O(1)��Al(2) bond did not decompose because of a short time of dynamics simulation. Thus, it could be predicted that O(1)��Al(2) would dissociate and then adsorbed CO molecule would generate after longer dynamics simulation time. There was free Cl(2) atom generated by dissociating of the new Al(2)��Cl(2) bond after 1 ps dynamics simulation but chlorine element could not be detected from gaseous product in process of carbothermic reduction and chloride to produce aluminum [25]. So, it could be deduced that free Cl(2) atom would react with Al(2) atom dissociated from the absorbed AlO molecule again, and Al(3) atom also interacted with Cl atom of other AlCl2 molecule, generating new absorbed AlCl molecules. we couldfurther predict that absorbed CO and AlCl molecules would escape from C(001) crystal plane, forming free CO and AlCl molecules after a longer period of dynamics simulation time under conditions of temperature 1760 K and the pressure of 60 Pa.
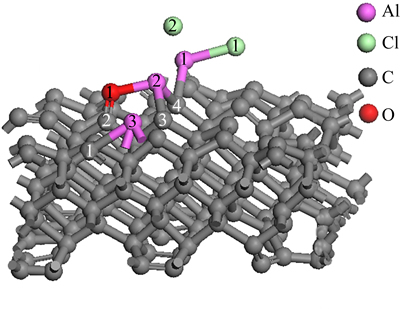
Fig. 6 structure of AlCl2 and Al2O on C(001) surface after 1 ps dynamics simulation
Figure 7 represents the structure and PDOS (partial density of states) of the free AlCl molecule after optimization. The bond length of the free AlCl molecule 2.15144 was approximate equal to that of Al(1)��Cl(1) 2.15813 after 1 ps dynamics simulation. Thereby, it could be predicted that the bond lengths of AlCl molecules adsorbed on C(001) surface would decrease to 2.15144 and then escape from the C(001) surface after a longer period of dynamics simulation time. From thePDOS of AlCl, including six regions, we noticed that nearby the 0 eV region, the PDOS was mainly from Al 3s orbital, and was mainly contributed by Cl 3p state nearby the -2 eV region. In the range from -4 to -3 eV and nearby the 6 eV, the PDOS was mainly contributed by the Al 3p orbital. The PDOS was mainly from Cl 3s state between -14 and -13 eV.
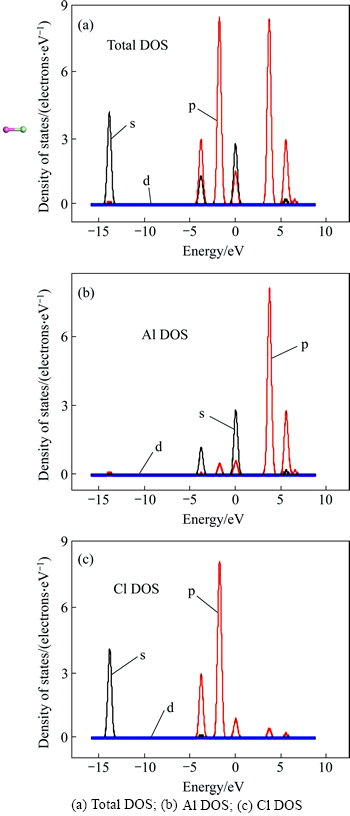
Fig. 7 Structure and PDOS of AlCl molecule:
Figure 8 shows the PDOS results of adsorbed Al(1)��Cl(1) molecule, which was formed by Al2O with AlCl2 and C after 1 ps dynamics simulation, and except a significantly broadened range at -7.5-2 eV, the PDOS peaks of adsorbed Al(1)��Cl(1) molecule in Fig. 8 is roughly the same as the PDOS of free AlCl molecule inFig. 7. therefore, we could further draw conclusion that under conditions of temperature 1760 K and the pressure of 60 Pa, the reaction of Al2O and AlCl2 with C could be carried out through successive steps: firstly, adsorbed AlCl molecules were generated; secondly, these AlCl molecules would escape from C surface and exist in the form of free AlCl molecules. By comparing the PDOS of Al(1), Cl(1) in Fig. 8 with Al, Cl in Fig. 7, it can be found there was a significant negative shift of the Cl(1) 3s peaks and Cl(1) 3p orbital was significantly broadened at -7.5-2 eV and Al(1) 3s, 3p orbitals were significantly broadened at -9-2 eV in Fig. 8.

Fig. 8 PDOS of adsorbed Al(1)��Cl(1) molecule after 1 ps dynamics simulation:
Figure 9 represents the structure of AlCl2 molecule and Al2O molecule on C(001) crystal plane after 1.25 ps dynamics simulation. By comparing Fig. 9 and Fig. 6, it could be found that adsorbed CO molecule and Al(2)��Cl(2) molecule were formed after more dynamics simulation time in this system. The result, after 1.25 ps dynamics simulation, indicated that the previous conjectures were reasonable and correct. The previously experimental result showed that AlCl and CO were gaseous product of carbothermal reduction-chlorination of alumina before AlCl disproportionation [25]. Therefore, it obviously meant that adsorbed AlCl molecules and CO molecule would escape from C(001) surface after a longer period of dynamics simulation time.
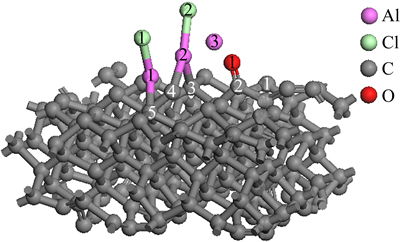
Fig. 9 structure of AlCl2 and Al2O on C(001) surface after 1.25 ps dynamics simulation
Combined with the interaction and dynamic simulation, the mechanism of the reaction among Al2O(g), AlCl2(g) and carbon could be represented by the following reactions at 1760 K and the pressure of 60 Pa (* shows adsorbed state on C surface )
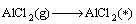 (12)
(12)
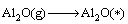 (13)
(13)
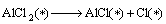 (14)
(14)
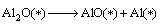 (15)
(15)
 (16)
(16)
 (17)
(17)
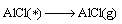 (18)
(18)
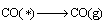 (19)
(19)
4 Conclusions
1) The spontaneous reaction temperatures calculated were 920.22, 1014.81, 1131.08, 1277.44, 1467.31 K under the pressure of 10, 102, 103, 104 and 105 Pa, respectively. Thus, the reaction could be performed under conditions of temperature 1760 K and under the pressure of 60 Pa.
2) AlCl2 adsorbed on substrate C(001) surface had been dissociated in the presence of Al2O and then new chemical bond had been formed between Al atom in Al2O and Cl atom from one of Al��Cl bonds in AlCl2 molecule dissociated. However, Al2O adsorbed on C(001) crystal surface did not decompose before dynamics simulation.
3) Adsorbed AlCl molecules had been generated and CO molecule would be formed in Al2O��AlCl2��C system, but they didn��t escape after 1.25 ps dynamics simulation, and they would escape from C surface after a longer period of dynamics simulation time, namely, the reaction of Al2O and AlCl2 with C could be carried out under given constraint condition, corresponding to the result of thermodynamic calculation.
References
[1] MURRY J P. Aluminum production using high-temperature solar process heat [J]. Solar Energy, 1999, 66(2): 133-142.
[2] NAMBOOTHIRI S, TAYLOR M P, CHEN J J J, HYLANDL M M, COOKSEY M. Aluminium production options with a focus on the use of a hydrogen anode: a review [J]. Asia-Pacific Journal of Chemical Engineering, 2007, 2(5): 442-447.
[3] BRUNO M J. Aluminium carbothermic technology comparison to Hall-Heroult process [C]// Light Metals-Warrendale-Proceedings. TMS, 2003: 395-400.
[4] MYKLEBUST H, RUNDE R. Greenhouse gas emissions from aluminum carbothermic technology compared to hall-heroult technology [J]. Light Metals, 2005: 519-522.
[5] CHOATE W, GREEN J. Technoeconomic assessment of the carbothermic reduction process for aluminum production [J]. Essential Readings in Light Metals: Aluminum Reduction Technology, 2006, 2: 1070-1075.
[6] KRUESI M, GALVEZ M E, HALMANN M, STEINFELD A. Solar aluminum production by vacuum: carbothermic reduction of alumina-thermodynamic and experimental analyses [J]. Metallurgical and Materials Transactions B, 2011, 42(1): 254-260.
[7] VISHNEVESTSKY I, ZVI R H, EPSTEIN M, BARAK S, RUBIN R. Solar carboreduction of alumina under vacuum [J]. Journal of the Minerals Metals & Materials Society, 2013, 65(12): 1721-1732.
[8] YU Qing-chun, YUAN Hai-bin, ZHU Fu-long, ZHANG Han, WANG Chen, LIU Da-chun, YANG bin. Carbothermic reduction of alumina with carbon in vacuum [J]. Journal of Central South University, 2012, 19(7): 1813-1816.
[9] FENG Yue-bin, YU Qing-chun, YANG Bin, DAI Yong-nian. Extraction of aluminum from alumina by disproportionation process of AlCl in vacuum [J]. Transactions of Nonferrous Metals Society of China, 2013, 23(9): 2781-2785.
[10] FENG Yue-bin, YANG Bin, DAI Yong-nian. Carbothermal recuction-chlorination- disproportionation of alumina in vacuum [J]. Transactions of Nonferrous Metals Society of China, 2012, 22(1): 215-221.
[11] DUAN Shao-fei, CHEN Xiu-min, YANG Bin, YU Qing-chun, XU Bao-qiang, LIU Da-chun. Calculation of interaction of AlCl, AlCl2 and AlCl3 on Al4C3 (001) Al4CO4 (001) and Al2CO (001) planes [J]. Journal of Central South University, 2015, 22(1): 43-58.
[12] YE Da-lun. Practical handbook of the thermodynamic data for inorganic compounds [M]. Beijing: Metallurgical Industry Press, 2002. (in Chinese)
[13] LANDOLT B. Thermodynamic properties of inorganic material [M]. Berlin: Springer-Verlag, 1999.
[14] BONNIE J M. Thermodynamic data for fifty reference elements [M]. Cleveland: National Aeronautics and Space Administration, 1993.
[15] BARIN I, PLATZKI G. Thermochemical data of pure substances, part 1 [M]. Weinheim: VCH Verlags Gesellschaft, 1993.
[16] PFROMMER B G, COTO M, LOUIE S G, COHEN M L. Relaxation of crystals with the quasi-newton method [J]. Journal of Computational Physics, 1997, 131(1): 133-140.
[17] BECKE A D. Density functional calculations of molecular-bond energies [J]. The Journal of Chemical Physics, 1986, 84(8): 4524-4529.
[18] PERDEW J P, WANG Y. Accurate and simple density functional for the electronic exchange energy: generalized gradient approximation [J]. Physical Review B, 1986, 33(12): 8800-8802.
[19] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalues formalism [J]. Physical Review B, 1990, 41(11): 7892-7895.
[20] FRANCIS G P, PAYNE M C. Finite basis set corrections to total energy pseudopotential calculations [J]. Journal of Physics, 1990, 2(19): 4395-4404.
[21] ANDERSEN H C. Molecular dynamics at constant pressure and/or temperature [J]. Journal of Computational Physics, 1980, 72(4): 2384-2393.
[22] ANISIMOV V, ZAANEN J, ANDERSEN O K. Band theory and mott insulators: Hubbard U instead of stoner [J]. Physical Review B, 1991, 44(3): 943-954.
[23] NOS S. A molecular dynamics method for simulations in the canonical ensemble [J]. Molecular Physics, 1984, 52(2): 255-268.
S. A molecular dynamics method for simulations in the canonical ensemble [J]. Molecular Physics, 1984, 52(2): 255-268.
[24] MARX D, HUTTER J. Ab initio molecular dynamics: Basic theory and advanced methods [M]. London: Cambridge University Press, 2009.
[25] YUAN Hai-bin, FENG Yue-bin, YANG Bin, YU Qing-chun, XU Bao-qiang, WANG Peng-cheng, DAI Yong-nian. Thermal behavior of alumina in process of carbothermic reduction and chloride to produce aluminum [J]. The Chinese Journal of Nonferrous Metals, 2010, 20(4): 777-783. (in Chinese)
(Edited by YANG Hua)
Foundation item: Projects(51104078, 51264023) supported by the National Natural Science Foundation of China; Project(2010CD022) supported by Yunnan Province Applied Basic Research Fund, China; Project(IRT1250) supported by the Program for Innovative Research Team in University of Ministry of Education of China; Project(U1202271) supported by the National Natural Science Foundation of China- Yunnan United fund; Project(KKZ3201252020) supported by Kunming University of Science and Technology for Talent Training, China
Received date: 2014-12-04; Accepted date: 2015-04-10
Corresponding author: CHEN Xiu-min, Professor, PhD; Tel: +86-871-5107208; E-mail: chenxiumin9@hotmail.com

