J. Cent. South Univ. Technol. (2009) 16: 0223-0229
DOI: 10.1007/s11771-009-0038-y

Acidophilic bacterial community reflecting pollution level of sulphide mine impacted by acid mine drainage
WAN Min-xi(万民熙)1, YANG Yu(杨 宇)1, 2, QIU Guan-zhou(邱冠周)1, 2,
XU Ai-ling(徐爱玲)1, QIAN Lin(钱 林)1, HUANG Zhi-ying(黄芝英)1, XIA Jin-lan(夏金兰)1, 2
(1. School of Resources Processing and Bioengineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Biometallurgy of Ministry of Education, Central South University, Changsha 410083, China)
Abstract: To reveal the impact of mining on bacterial ecology around mining area, bacterial community and geochemical characteristics about Dabaoshan Mine (Guangdong Province, China) were studied. By amplified ribosomal DNA restriction analysis and phylogenetic analysis, it is found that mining pollution greatly impacts the bacterial ecology and makes the habitat type of polluted environments close to acid mine drainage (AMD) ecology. The polluted environment is acidified so greatly that neutrophil and alkaliphilic microbes are massively dead and decomposed. It provided organic matters that can make Acidiphilium sp. rapidly grow and become the most bacterial species in this niche. Furthermore, Acidithiobacillus ferrooxidans and Leptospirillum sp. are also present in this niche. The amount of Leptospirillum sp. is far more than that of Acidithiobacillus ferrooxidans, which indicates that the concentration of toxic ions is very high. The conclusions of biogeochemical analysis and microbiological monitor are identical. Moreover, because the growth of Acidithiobacillus ferrooxidans and Leptospirillum sp. depends on ferrous iron or inorganic redox sulfur compounds which can be supplied by continual AMD, their presence indicates that AMD still flows into the site. And the area is closer to the outfalls of AMD, their biomasses would be more. So the distinction of their biomasses among different areas can help us to find the effluent route of AMD.
Key words: bacterial community; acid mine drainage (AMD); Acidithiobacillus ferrooxidans; Leptospirillum sp.
1 Introduction
Above 95% of the first energy, 80% of the original material in the industry and above 70% of the means of production in the agriculture all come from the mineral industry. However, the exploitation of the mineral resources greatly influences the environment.
Subsurface mining often progresses below the water table, in which water must be constantly pumped out of the mine in order to prevent flooding. And tailings piles or ponds in all mining may also be a source of acid mine drainage (AMD) [1]. Spontaneous oxidation of metal sulfides and elemental sulfur to sulfuric acid results in environmental acidification and toxic metal migration [2]. Compared with unpolluted streams, mines draining water and mine spoils are therefore often acidic, and frequently contain high concentrations of aluminum and various heavy metals, such as copper, zinc and manganese [3]. Therefore, mining results in the contamination of ground and/or surface waters, which represent local ecological problems [1].
Up to date, researches about the animal and plant ecosystem influenced mining are numerous; however, bacterial ecology impacted by mine drainage around mining area is different from that in AMD and is seldom studied. The aims of studies about bacterial ecology in AMD usually are to understand the structure of microbial communities in different AMDs, and to present a more complete picture of microbially mediated AMD production [4-6], however, investigations about bacterial ecology impacted by mine drainage can reveal the impact of mining on bacterial ecology around mining area.
In this study, bacterial community and geochemical characteristics of area impacted by mine drainage about Dabaoshan Mine (Guangdong Province, China) were investigated. The genomic DNA of the bacterial community was extracted. Subsequently, Amplified Ribosomal DNA Restriction Analysis (ARDRA) was used to analyze the bacterial communities.
2 Materials and methods
2.1 Study site, sample collection and physicochemical analyses
Dabaoshan Mine (24.5? N, 113.7? E) is located in the mountainous area of the northern part of Guangdong Province, China. It is a large, multimetallic mineral deposit: the top of the main ore body appears to be a limonite body, while the lower body contains copper-sulfur compounds and associated tungsten, bismuth, molybdenum, gold, and silver metal ores. As in the case of most metal deposits, the strata contain pyrite (FeS2) and various other metal sulfide ores. The sample was collected from a pool near the drainage of Dabaoshan Mine. The water sample was filtered through 0.2 ?m nylon filters (Jinjing, China) using a vacuum pump, and then immediately transferred into anaerobic jar. The sediment on the filters was collected and maintained at -20 ℃.
The filtrate of sample was used for chemical analysis. Physicochemical analysis of the water samples are performed at the Testing Center of Central South University. Flame atomic absorption spectrometry was used for metal ion measurements, and pH was measured using a pH meter (Leici, China.).
2.2 DNA extraction and purification
The microorganisms of sample were isolated from the sediment obtained after filtration of the samples through 0.2 ?m nylon filters. The genomic DNA of the bulk community was extracted from 5 g of sediment following the protocols described by HURT et al [7]. And the quality of genomic DNA was analyzed by agarose gel electrophoresis and purified using a Wizard DNA clean-up kit (Promega).
2.3 16S rRNA gene amplification and cloning
Two primers were used to amplify approximately 1 300 bp of a consensus 16S rRNA gene fragment: forward primer 63F (5’-CAGGCCTAACACATGCAA- GTC-3’) and reverse primer 1387R (5’-GGGCGGWGT- GTACAAGGC-3’) [8].
The polymerase chair reaction (PCR) mixtures (50 μL) contained 10 pmol of each appropriate primer, 2 μL of genomic DNA (about 0.1 ?g/?L), 5 μL of 10×dNTP (2 mmol/L each), 5 μL of 10×PCR buffer, and 0.5 U of AmpliTaq Gold DNA polymerase (Perkin Elmer). Thermal cycling was carried out using a Whatman Biometra T1 thermocycler programmed for an initial step of 5 min at 94 ℃, followed by 30 cycles of 45 s at 94 ℃, 45 s at 65 ℃, 90 s at 72 ℃, and then 7 min at 72 ℃. The PCR products were visualized by 1.0% low-melting-point agarose gel electrophoresis, and purified using Wizard DNA Clean-Up Kit (Promega).
The purified 16S rRNA gene fragments were ligated into the vector pCR2.1 TOPO and then transformed into E. coli TOP10F’ competent cells according to the manufacturers’ instructions (Invitrogen). Recombinants were identified based on blue-white screening, and grown overnight in Luria-Bertani (LB) agar plates containing appropriate amounts of ampicillin, X-gal and IPTG at 37 ℃. Next, white colonies from the library were randomly selected, and the recombinant plasmids containing 16S rRNA gene fragments were reamplified by PCR using the vector primers M13F (5’-GTAAAA- CGACGGCCAGTG-3’) and M13R (5’-GGAAACAGC- TATGACCATG-3’). The PCR mixtures (50 μL) also contained 10 pmol of each appropriate primer, 2 μL of plasmid DNA (about 0.1 μg/μL), 5 μL of 10×dNTP (2 mmol/L each), 5 μL of 10×PCR buffer, and 0.5 U of AmpliTaq Gold DNA polymerase (Perkin Elmer). Thermal cycling was carried out using a Whatman Biometra T1 Thermocycler programmed for an initial step of 3 min at 94 ℃, followed by 30 cycles of 45 s at 94 ℃, 45 s at 55 ℃, 90 s at 72 ℃, and then 7 min at 72 ℃.
2.4 Amplified ribosomal DNA restriction analysis (ARDRA)
The amplified rRNA PCR products of the correct size (approximately 1.3 kb) were digested overnight at 37 ℃ using Hin6I and MspI (Fermentas). The resulting ARDRA products were separated by gel electrophoresis in 3.0% agarose. The ARDRA patterns were visualized by ultraviolet (UV) excitation. Jaccard coefficients were computed for all pairwise comparisons of ARDRA banding patterns, and dendrograms were constructed by the unweighted pair group mean average method using the Molecular Analyst version 1.1 software (Bio-Rad). The ARDRA banding patterns that were identified were grouped into an operational taxonomic unit (OTU), and a representative clone was selected for nucleotide sequence determination in each OTU.
2.5 Sequencing and phylogenetic analysis
After selecting clones with different ARDRA patterns, related clones were sequenced by Sunbiotech (Beijing). Sequence identification was initially estimated at the BLASTN facility of the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/ BLAST). The initial phylogenetic trees were based on all available sequences and constructed using the DNA distance neighbor-joining program with Felsenstein correction in ARB (from Latin arbor, tree) [9]. After appropriate subsets of 16S rRNA gene sequences were selected, analyzed, and aligned using CLUSTAL-X (version 1.8), the final phylogenetic trees were generated based on the initial phylogenetic results.
2.6 Statistical analysis
Because the reciprocal of Simpson’s index (1/D) is of good discriminating ability, and is used widely in ecological studies, it was chosen to characterize the microbial communities in this study. The use of 1/D instead of the original formulation of Simpson’s index ensures that an increase in the reciprocal index reflects an increase in diversity [10]. The equation of Simpson’s index is:
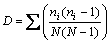
For the full community D is simply 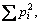 where pi is the proportion of clones in the ith OTU. However, for finite sample ni represents the number of clones in the ith OTU, and N represents the sum of clones.
where pi is the proportion of clones in the ith OTU. However, for finite sample ni represents the number of clones in the ith OTU, and N represents the sum of clones.
Rarefaction analysis was performed using the SigmaPlot version 8.0 software. The exponential model y=a[1-exp(-bx)] was applied using SigmaPlot to fit the clone distribution data.
3 Results
3.1 Biogeochemical properties of sample
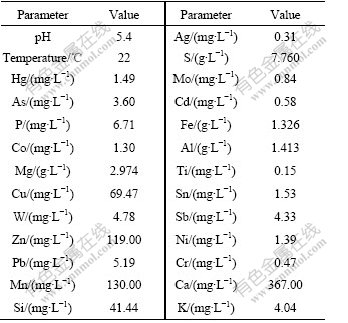
The biogeochemical properties of the water sample are listed in Table 1. Compared with Surface Water Quality Standards (GB3838―2002), Cu (69.47 mg/L), Zn (119.00 mg/L), As (3.60 mg/L), Hg (1.49 mg/L) and Cd (0.58 mg/L) are 69, 59, 36, 1 490, 58 times than standard values, respectively.
Table 1 Biogeochemical properties of sample
3.2 ARDRA of 16S rRNA clone library
In this study, clones containing partial 16S rRNA gene inserts were obtained by the direct cloning of PCR products. 91 Clones for the 16S rRNA gene clone library were analyzed by ARDRA. And the representative ARDRA patterns of the sample are shown in Fig.1. A total of 32 OTUs were obtained from these ARDRA patterns. Percentage of each clone is shown in Fig.2. Two OTUs (Nos. 8 and 18) are the most dominant; however, OTUs (Nos. 2, 3, 5, 6, 7, 11, 13, 14, 16, 24, 26, 30, 31 and 32) are the least.
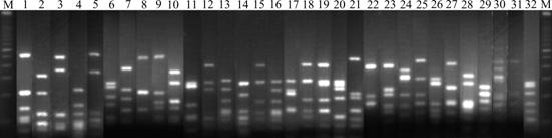
Fig.1 Representative ARDRA profiles of 16S rRNA fragment amplified from water sample (1, 2, …, 32 represent clone 1, clone 2, …, clone 32, respectively; M represents 100 bp DNA ladder plus)
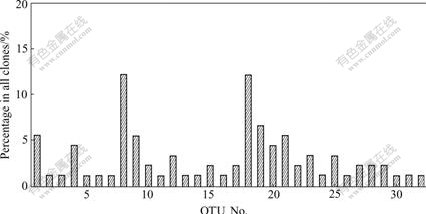
Fig.2 Percentage of clones in water sample
3.3 Sequence data and phylogenetic analysis of 16S rRNA gene
To determine the phylogenetic diversity, representative 16S rRNA clones were sequenced. Comparative sequence analysis was conducted on nucleotide sequences (on average, 1 310 comparable positions for 16S rRNA gene). The major sequences of clones differed by less than 5% from those in current databases. The phylogenetic relationships of all clones were established with a bootstrap neighbor-joining method using the sequences from all known and candidate divisions (Fig.3). These sequences fell into 4 main putative phylogenetic divisions, Acidobacteria, Nitrospira, α-Proteobacteria and γ-Proteobacteria, which amounted to OTUs of 2, 5, 11 and 14 respectively. Clones were affiliated with γ-Proteobacteria (46.2%), α-Proteobacteria (39.5%), Nitrospira (7.7%) and Acidobacteria (6.6%). The most bacteria were Acidiphilium sp. (25.3%), Leptospirillum ferrooxidans (11.0%) and Acidithiobacillus ferrooxidans (1.1%).
3.4 Statistical analysis
Calculations of bacterial diversity revealed that the diversity of bacterial ecology (1/D=22.02) impacted by Mine Drainage was very low. Rarefaction curves (plots of the cumulative number of OTUs as a function of clone number) indicated that the majority of the OTUs in the sample were detected (shown in Fig.4). This suggests that the level of analysis is sufficient to detect community diversity and infer the distribution within these communities.
4 Discussion
To quantitatively measure diversity in the sample, the inverse of Simpson’s index (1/D) was used, which is sensitive to the level of dominance in a community. The reciprocal of Simpson’s index effectively proved that the diversity of bacterial ecology (1/D=22.02) impacted by mine drainage was much lower than unpolluted environment, such as surface soils, marine sediments and so on. ZHOU et al [10] showed that 1/D values in surface soils ranged from 507 to 27 331, while RAVENSCHLAG et al [11] demonstrated that bacterial diversity in permanently cold marine sediments was high. The 1/D value of actinobacterial diversity was 51, even 5 to 12 cm below the sea floor at a depth of 3.814 km [12]. The environment polluted by mining examined in the present study exhibited very low microbial diversity indices because of high ion concentrations. And the value was closer with AMD’s (1/D: 2.51-18.42) than that of unpolluted environment. The results were noteworthy that mining pollution could greatly impact the bacterial ecology and make bacterial communities of polluted environments close to AMD communities.
In AMD ecology, Acidobacteria was a genus of very important microbes, which performs an indispensable function in the formation of AMD. Acidobacteria existed in relatively moderate AMD environments (20-37 ℃ and pH of 3.0-6.0). Some analyses suggested that this group was limited to environments of high pH (pH>1.4) [13]. Acidobacteria found in the sample showed that AMD could lead the habitat type of polluted environments to close AMD type.
A heterotrophic bacterial species, Acidiphilium sp., was found in the site. Acidiphilium sp. is considered able to adapt to temperatures ranging from 17 to 45 ℃, and to pH values ranging from 1.5 to 6.0 [2]. PECCIA et al [14] suggested that there might be a mutualistic relationship between Acidithiobacillus sp. and Acidiphilium sp. Acidiphilium sp. was reported along with iron- and sulfur-oxidizing chemolithoautotrophs in extreme acidic environments [15]. These microorganisms may play a critical role in such ecosystems. For example, some studies proposed that Acidiphilium sp. could remove organic toxic compounds for Leptospirillum and Acidithiobacillus [16]. Meanwhile, the physiological and ecological capabilities of Acidiphilium sp., which can utilize a variety of substrates and reduce ferric iron in the absence of oxygen, also indicate its important ecological role in AMD, especially in the turnover of iron at oxic-anoxic interfaces [17]. In the study, Acidiphilium sp. was the most bacterium. The pollution could cause the environment to acidify so greatly that neutrophil and alkaliphilic microbes were massively dead and decomposed. It provided organic matters that could make Acidiphilium sp. rapidly grow and become the most bacterial species in the niche.
A. ferrooxidans and L. ferrooxidans are chemolithotrophic prokaryotes that use ferrous iron as a source of energy [18] and are widely considered to be the microorganisms that control the rate of generation of AMD and have been used as model-leaching environ- ments [19-20]. Also, Acidithiobacillus ferrooxidans can use inorganic reduced sulfur compounds as a source of energy. To date, Leptospirillum isolates and environmentally-derived clones cluster within one of three phylogenetically distinct groups [21]. The three groups were as follows: L. ferrooxidans (groupⅠ) [22], L. ferriphilum (group Ⅱ) [23] and Leptospirillum (group Ⅲ). Group Ⅲ has only been detected via clone library analysis of Iron Mountain bacterial communities [13, 21]. In this study, Leptospirillum group Ⅰsequences were discovered, and were dominant in Leptospirillum groups. Only one sequence was affiliated with group Ⅲ, no one was affiliated with the group Ⅱ. L. ferrooxidans is reported to grow in an optimal pH range of 1.6-2.0 [23]. A. ferrooxidans with the greatest abundance (>30%) at moderate temperature (2.5-20 ℃) and pH (1.5-2.3) were produced in sites that were peripheral to primary acid-generating sites. And they play important role in dissolution of minerals, and are often isolated from AMD at low pH and high ionic strength of the habitat and brings water/soil pollution [19-20]. When AMD flows into ponds and rivers around mines, the bacteria could be brought [24]. In the pond, Acidithiobacillus ferrooxidans and Leptospirillum sp. were 1.1% and 11.0% in all of bandings, although they were badly less in the pond than in AMD, which indicated that the pond was badly polluted. Because Leptospirillum sp. had a strong tolerance to toxic ions, the amount of Leptospirillum sp. was far more than that of Acidithiobacillus ferrooxidans, which indicated that the concentration of toxic ions might be very high in the niche. Compared with Surface Water Quality Standards (GB3838―2002), the results of biogeochemical analysis and microbiological monitor were identical.
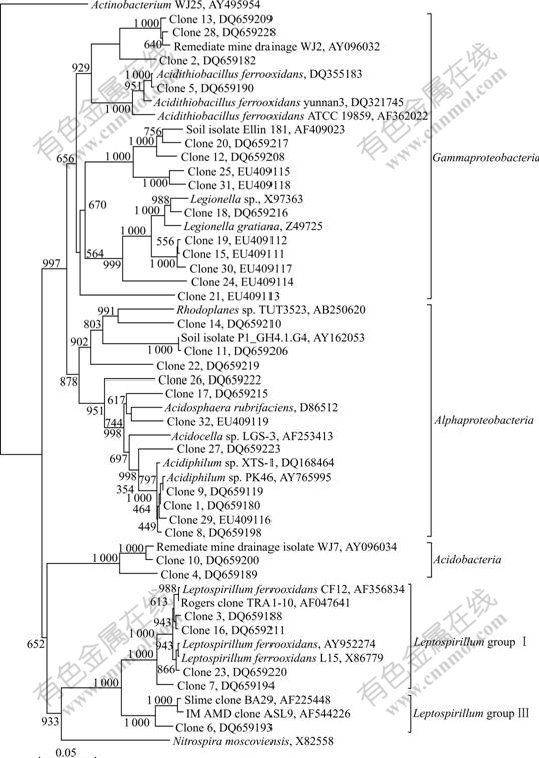
Fig.3 Phylogenetic relationship of partial 16S rRNA sequences generated in this study (Scale bar represents the number of changes per base position. Numbers at tree nodes represent the number of times the topology to the right of the node is recovered in 1 000 bootstrap resamplings. The 16S rRNA gene sequences of Clone 1 to Clone 32 described in this study are submitted to GenBank, and their accession numbers are shown in Fig.3)
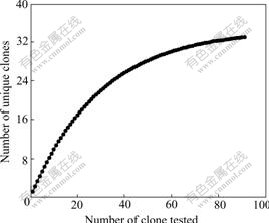
Fig.4 Rarefaction curves for different ARDRA patterns expected of all clones used in this study
Moreover, because the growth of Acidithiobacillus ferrooxidans and Leptospirillum sp. depend on ferrous iron or inorganic redox sulfur compounds which must be supplied by continual AMD, their presence indicated that AMD still flowed into the pond. And the result showed that the area was closer to the outfalls of AMD, their biomasses would be more. So the distinction of their biomasses in different areas can help us to find the effluent route of AMD.
5 Conclusions
(1) Mining pollution could greatly impact the bacterial ecology and make the habitat type of polluted environments close to AMD’s.
(2) The pollution could cause the environment to acidify so greatly that neutrophil and alkaliphilic microbes were massively dead and decomposed. It provided organic matters could make Acidiphilium sp., a heterotrophic acidophilic bacterium, rapidly grow and become the most bacterial species in the niche.
(3) The area was closer to the outfalls of AMD, and the biomasses of Acidithiobacillus ferrooxidans and Leptospirillum sp. would be more. The distinction of their biomasses in different areas can help us to find the effluent route of AMD.
References
[1] JOHNSON D B, HALLBERG K B. The microbiology of acidic mine waters [J]. Research in Microbiology, 2003, 154(7): 466-473.
[2] BAKER B J, BANFIELD J F. Microbial communities in acid mine drainage [J]. Fems Microbiology Ecology, 2003, 44(2): 139-152.
[3] OKABAYASHI A, WAKAI S, KANAO T, SUGIO T, KAMIMURA K. Diversity of 16S ribosomal DNA-defined bacterial population in acid rock drainage from Japanese pyrite mine [J]. Journal of Bioscience and Bioengineering, 2005, 100(6): 644-652.
[4] YANG Yu, SHI Wu-yang, WAN Min-xi, ZHANG Yan-fei, ZOU Li-hong, HUANG Ju-fang, QIU Guan-zhou, LIU Xue-duan. Diversity of bacterial communities in acid mine drainage from the Shen-bu copper mine, Gansu Province, China [J]. Electronic Journal of Biotechnology, 2008, 11(1): 108-117.
[5] ZHOU Hong-bo, LIU Xi, FU Bo, QIU Guan-zhou, HUO Qiang, ZENG Wei-min, LIU Jian-she, CHEN Xin-hua. Isolation and characterization of acidithiobacillus caldus from several typical environments in China [J]. Journal of Central South University of Technology, 2007, 14(2): 163-169.
[6] YIN Hua-qun, QIU Guan-zhou, WANG Dian-zuo, CAO Lin-hui, DAI Zhi-min, WANG Jie-wei, LIU Xue-duan. Comparison of microbial communities in three different mine drainages and their bioleaching efficiencies to low grade of chalcopyrite [J]. Journal of Central South University of Technology, 2007, 14(4): 460-466.
[7] HURT R A, QIU X Y, WU L Y, ROH Y, PALUMBO A V, TIEDJE J M, ZHOU J Z. Simultaneous recovery of RNA and DNA from soils and sediments [J]. Applied and Environmental Microbiology, 2001, 67(10): 4495-4503.
[8] MARCHESI J R, SATO T, WEIGHTMAN A J, MARTIN T A, FRY J C, HIOM S J, WADE W G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA [J]. Applied and Environmental Microbiology, 1998, 64(2): 795-799.
[9] SMITH S W, OVERBEEK R., WOESE C R., GILBERT W, GILLEVET P M. The genetic data environment an expandable gui for multiple sequence-analysis [J]. Computer Applications in the Biosciences, 1994, 10(6): 671-675.
[10] ZHOU J Z, XIA B C, TREVES D S, WU L Y, MARSH T L, O'NEILL R V, PALUMBO A V, TIEDJE J M. Spatial and resource factors influencing high microbial diversity in soil [J]. Applied and Environmental Microbiology, 2002, 68(1): 326-334.
[11] RAVENSCHLAG K, SAHM K, PERNTHALER J, AMANN R. High bacterial diversity in permanently cold marine sediments [J]. Applied and Environmental Microbiology, 1999, 65(9): 3982-3989.
[12] STACH J E M, MALDONADO L A, MASSON D G, WARD A C, GOODFELLOW M, BULL A T. Statistical approaches for estimating actinobacterial diverity in marine sediments [J]. Applied and Environmental Microbiology, 2003, 69(10): 6189-6200.
[13] DRUSCHEL G K, BAKER B J, GIHRING T M, BANFIELD J F. Acid mine drainage biogeochemistry at Iron Mountain, California [J]. Geochemical Transactions, 2004, 5(2): 13-32.
[14] PECCIA J, MARCHAND E A, SILVERSTEIN J, HERNANDEZ M. Development and application of small-subunit rRNA probes for assessment of selected Thiobacillus species and members of the genus Acidiphilium [J]. Applied and Environmental Microbiology, 2000, 66(7): 3065-3072.
[15] GONZALEZ-TORIL E, LLOBET-BROSSA E, CASAMAYOR E O, AMANN R, AMILS R. Microbial ecology of an extreme acidic environment, the Tinto River [J]. Applied and Environmental Microbiology, 2003, 69(8): 4853-4865.
[16] JOHNSON D B. Selective solid media for isolating and enumerating acidophilic bacteria [J]. Journal of Microbiological Methods, 1995, 23(2): 205-218.
[17] KUSEL K, DORSCH T, ACKER G, STACKEBRANDT E. Microbial reduction of Fe(Ⅲ) in acidic sediments: Isolation of Acidiphilium cryptum JF-5 capable of coupling the reduction of Fe(Ⅲ) to the oxidation of glucose [J]. Applied and Environmental Microbiology, 1999, 65(8): 3633-3640.
[18] JOHNSON D B, ROLFE S, HALLBERG K B, IVERSEN E. Isolation and phylogenetic characterization of acidophilic microorganisms indigenous to acidic drainage waters at an abandoned Norwegian copper mine [J]. Environmental Microbiology, 2001, 3(10): 630-637.
[19] ROHWERDER T, GEHRKE T, KINZLER K, SAND W. Bioleaching review part A: Progress in bioleaching: fundamentals and mechanisms of bacterial metal sulfide oxidation [J]. Applied Microbiology and Biotechnology, 2003, 63(3): 239-248.
[20] FOWLER T A, HOLMES P R, CRUNDWELL F K. Mechanism of pyrite dissolution in the presence of Thiobacillus ferrooxidans [J]. Applied and Environmental Microbiology, 1999, 65(7): 2987-2993.
[21] BOND P L, SMRIGA S P, BANFIELD J F. Phylogeny of micro- organisms populating a thick, subaerial, predominantly lithotrophic biofilm at an extreme acid mine drainage site [J]. Applied and Environmental Microbiology, 2000, 66(9): 3842-3849.
[22] HIPPE H. Leptospirillum gen. nov (ex Markosyan 1972), nom. rev., including Leptospirillum ferrooxidans sp nov (ex Markosyan 1972), nom. rev. and Leptospirillum thermoferrooxidans sp nov (Golovacheva et al. 1992) [J]. International Journal of Systematic and Evolutionary Microbiology, 2000, 50: 501-503.
[23] CORAM N J, RAWLINGS D E. Molecular relationship between two groups of the genus Leptospirillum and the finding that Leptosphillum ferriphilum sp nov dominates South African commercial biooxidation tanks that operate at 40 ℃ [J]. Applied and Environmental Microbiology, 2002, 68(2): 838-845.
[24] YANG Yu, WAN Min-xi, SHI Wu-yang, PENG Hong, QIU Guang-zhou, ZHOU Ji-zhong, LIU Xue-duan. Bacterial diversity and community structure in acid mine drainage from Dabaoshan Mine, China [J]. Aquatic Microbial Ecology, 2007, 47(2): 141-151.
Foundation item: Project(50621063) supported by the Science Fund for Creative Research Groups of China; Project(2004CB619201) supported by the Major State Basic Research Development Program of China
Received date: 2008-05-21; Accepted date: 2008-07-23
Corresponding author: QIU Guan-zhou, Professor, PhD; Tel: +86-731-8877216; E-mail: qgz@mail.csu.edu.cn
(Edited by YANG You-ping)

