������-���嵰���ڸ�ѡ����������õĵ绯ѧ�о�
��Դ�ڿ����й���ɫ����ѧ��(Ӣ�İ�)2013���9��
�������ߣ�Taki G��LER Kemal ��AHBUDAK Sevil ��ET��NKAYA ��nal AKDEM��R
����ҳ�룺2766 - 2775
�ؼ��ʣ���������ĭ��ѡ���Ѱ��ף�������ԭ��Ӧ������
Key words��pyrite; froth flotation; ovalbumin; redox reactions; depression
ժ Ҫ�����嵰��(OVA)���׳Ƽ�������ף���һ�ֻ������������۵ı�����Լ������ķ��ӽṹ�к��е绯ѧ�����ϻ�������ѭ�����������������ѡʵ���о�OVA�ڻ����������������Ϊ������Ƽ����ڿ��ĵ�λ��Χ�ںͼ���pHֵ�����£��о��绯ѧ�������������ƺ������ʵ����á���OVA������ʱ���ɸ�������������λ�����´ﵽ����ֵ�������˵�λ��Χ���ɸ��Ի���������Fe�����������Լ���ˮ�����������͡����������Ե�λ��Χ��OVA-������������á�OVA������Ч�����ŵ�λ�ӻ�ԭ��λ����������λ����������Ŀ�·��λ�����ӣ�����Ҫ������OVA�������Ĺ���仯�Լ���·��λ�µ���ˮ��������ġ�OVA�����������е�������λ�´ﵽ����ֵ���ⱻ��Ϊ��������������OVA�Խ�����������ʽ�������ڽϸߵĵ�λ�����ָߵ�����Ч�����Լ���һֱ������ȥ��
Abstract: Ovalbumin (OVA), chicken egg albumin, is an environmentally friendly, non-toxic, cheap surface active agent. It has electrochemically active sulfhydryl residues in molecular structure. OVA adsorption on pyrite as an alternative depressant was investigated by cyclic voltammetry, FTIR spectroscopy and flotation. Tests were conducted in a wide potential range in alkaline pH to clarify the role of electrochemical condition on the adsorption mechanisms and the rate of depression. In the absence of OVA, the floatability reached its maxima around slightly oxidizing condition. Beyond the range, it decreased presumably due to the formation of Fe-oxyhydroxides together with the hydrophilic S-containing species. OVA-pyrite interaction occurred in the whole examined potential range. Depressing effect of OVA increased gradually from reducing to slightly oxidizing potential, open circuit potential (OCP) of pyrite, mainly due to weak conformational changes in OVA molecules together with the hydrophobic interaction around OCP. The rate of depression reached its maxima at moderately oxidizing potentials, which was referred to electrostatic attraction. This level was almost kept at higher potentials owing to OVA adsorption as metal-chelates.
Trans. Nonferrous Met. Soc. China 23(2013) 2766-2775
Taki  1, Kemal
1, Kemal  2, Sevil
2, Sevil  3,
3,  3
3
1. Department of Mining Engineering,  Sitki
Sitki  University, 48170, Turkey;
University, 48170, Turkey;
2. Department of Metallurgical and Materials Engineering, Cumhuriyet University, Sivas 58140, Turkey;
3. Department of Mining Engineering, Cumhuriyet University, Sivas 58140, Turkey
Received 28 November 2012; accepted 19 March 2013
Abstract: Ovalbumin (OVA), chicken egg albumin, is an environmentally friendly, non-toxic, cheap surface active agent. It has electrochemically active sulfhydryl residues in molecular structure. OVA adsorption on pyrite as an alternative depressant was investigated by cyclic voltammetry, FTIR spectroscopy and flotation. Tests were conducted in a wide potential range in alkaline pH to clarify the role of electrochemical condition on the adsorption mechanisms and the rate of depression. In the absence of OVA, the floatability reached its maxima around slightly oxidizing condition. Beyond the range, it decreased presumably due to the formation of Fe-oxyhydroxides together with the hydrophilic S-containing species. OVA-pyrite interaction occurred in the whole examined potential range. Depressing effect of OVA increased gradually from reducing to slightly oxidizing potential, open circuit potential (OCP) of pyrite, mainly due to weak conformational changes in OVA molecules together with the hydrophobic interaction around OCP. The rate of depression reached its maxima at moderately oxidizing potentials, which was referred to electrostatic attraction. This level was almost kept at higher potentials owing to OVA adsorption as metal-chelates.
Key words: pyrite; froth flotation; ovalbumin; redox reactions; depression
1 Introduction
Selectivity problem and concentrate dilution with gangue pyrite have always become the leading issues in flotation of complex sulfide ores mainly due to the similar electrochemical behavior of base metal sulfides. Although high conditioning pH, long conditioning time and extensive surface oxidation have been proposed to depress pyrite and to improve selectivity [1,2], using of additional chemicals has generally been a necessity to satisfy desired selectivity. Various inorganic agents (e.g., cyanide, hydroxides and sulfoxy species) are found to be quite effective in pyrite depression. They have been consumed in flotation plants in spite of many practical problems related to handle in addition to high cost, such as toxic, causing poisoning and several environmental hazards [3,4]. Nonetheless, consumption of inorganic depressants has been forced to increase because more complex and lower grade ores with acceptable recovery rates are needed to process to meet demand. Therefore, alternative environmentally friendly and cheap organic depressants have been researched to overcome problems regarding the consumption and environmental concerns.
Promising selectivity against pyrite has been observed both in laboratory and industrial scales with several organic agents, such as polysaccharides, bacteria and proteins. Polysaccharides (e.g. dextrin) are the commonly employed organic depressants. BICAK et al [4] explained their adsorption by hydrogen bonding, Bronsted acid�Cbase interaction, and electrostatic interaction based on the work conducted in a single mineral system. BOULTON et al [5] indicated that polyacrylamide polymers of low relative molecular mass depressed pyrite as a result of specific interaction with ferric ions/hydroxides. PATRA and NATARAJAN [6] exhibited that microbially (paenibacillus polymyxa) assisted flotation with xanthate was successful in selective removal of pyrite from a binary mixture of pyrite and sphalerite. They also reported that extracellular bacterial proteins, derived from paenibacillus polymyxa, improved floatability of galena and pyrite, and reduced hydrophobicity of chalcopyrite [7]. Bacterial adhesion was proposed to occur via functional groups of cell wall including carboxyl, amino and hydroxyl groups [8]. JIA et al [9] stated from FTIR and UVS spectra that the content of saccharide on the cell wall of bacteria was more than that of protein, and the adsorption on sulfide minerals occurred via some groups in saccharide of cell wall, such as O��H, C=O and C��O. CHEN et al [10] discussed the efficiency of depression related to functional groups of organic agents in single mineral system. They claimed that organic compounds only with single group of hydroxyl, carboxyl or amino, or with the combination of these groups in molecular structures, were ineffective in depressing pyrite, whereas sulfhydryl group (SH) gives good depressing performance to organic compounds due to electrochemical interaction between depressant and pyrite.
In this work, possible adsorption mechanisms of ovalbumin (OVA) on pyrite were discussed. OVA is a relative high molecular mass monomeric glycoprotein. OVA contains single chain of 385 amino acid residues linked to each other by C of carboxyl (��COOH) group and N of amine (��NH2) group of amino acids forming amide (peptide) bonds and backbone structure. Additionally, amino acids have sidechains which determine the type of them, and in consequence, type of protein. OVA has one disulfide (S��S) bond in cystine and four sulfhydryl (��SH) bonds in cysteine groups. Such sulfurous bonds make OVA electrochemically active in aqueous environment [11]. OVA has been known to be useful in cases of poisoning by heavy metals (such as Fe) as a chelator to heavy metals by trapping the metal ions within the sulfhydryl bonds of the protein [12]. However, electrochemistry of OVA adsorption on sulfide minerals is a virgin subject. Few researchers have engaged on the application of OVA in mineral processing, in which various cases other than electrochemical interaction of OVA with sulfide minerals have been taken up. Moreover, uses of different proteins in mineral processing besides OVA have commonly been studied. BASTRZYK et al [13] investigated the effect of OVA-surfactant interaction on magnesite rock flotation, and proposed that flotation efficiency was higher when both surfactant and OVA adsorbed on particle surface. REZWAN et al [14] stated that the adsorption process of bovine serum albumin (BSA) on SiO2 and AlOOH coated SiO2 particles depended on pH, and was chiefly dominated by electrostatic interaction between adsorbate and adsorbent. LIU et al [15] examined the cysteine adsorption on pyrite by FTIR spectroscopy technique, and revealed out that chemical interaction governed the entire adsorption process. ROJAS-CHAPANA and TRIBUTSCH [16] explained cysteine-pyrite interaction with disulfide formation between sulfhydryl groups of cysteine and free-SH groups from pyrite.
OVA can easily be suppliable, abundant and cheap agent. It has sulfhydryl and disulfide groups (S content is 0.3%-2.4%) that are expected to determine its behavior in electrochemically active aqueous environment [10, 15-17]. Therefore, this study was planned to elucidate the electrochemistry of pyrite-OVA interaction. Cyclic voltammetry (CV), FTIR spectroscopy and batch flotation tests were the tools in the experimental works. CV tests were applied to ascertain the effect of potential on pyrite-OVA interaction, while FTIR spectroscopy study was conducted to define the reaction products at certain polarization potentials. Flotation tests were performed to clarify the role of pulp potential and redox products on the floatability of pyrite in the absence and presence of OVA.
2 Experimental
Highly pure pyrite crystals (>98% FeS2), supplied from Artvin-Murgul deposits in Turkey, were used in experimental works. Characterization of mineral sample was performed with chemical analysis, metal microscopy, and XRD analysis by Bruker D8 Advance diffractometer. Major impurity of the sample was found to be quartz.
Experiments were carried out using pH 9.2 tetraborate (0.05 mol/L Na2B4O7��10H2O) buffer solution. Solution was deoxygenated by bubbling highly pure N2 (>99.998%) intensively for at least 15 min to reduce O2 content below 1��10-6, which was determined by YSI-5100 oxymeter. KBr-disc pellet method was applied in FTIR spectroscopy study. Spectroscopic grade KBr salt was used in pellet preparation as a non-absorbing matrix and background. OVA, supplied by Merck, was tested as organic modifying agent. It was high purity (98%) chicken egg albumin, soluble in cold water, and has a relative molecular mass of 45 g/mol.
Experimental works were performed in three electrode system cells, in which a calomel electrode and a Pt-foil were employed as reference and counter electrode, respectively. The working electrode for FTIR spectroscopy and flotation tests was a Pt-wire, while it was a shaped pyrite crystal in cyclic voltammetry (CV) setup. Construction of pyrite working electrode for CV tests is given elsewhere. All the potentials recorded were converted from saturated calomel electrode scale to standard hydrogen electrode (SHE) scale by adding 245 mV to the measured values. A Gamry PCI-750 potentiostat mounted into a computer was used for potential control. PHE-200 Physical Electrochemistry software of Gamry Co. was employed in the measure- ment of cyclic voltammograms, and the polarization of pyrite samples for FTIR spectroscopy and flotation tests. Cyclic voltammograms were recorded in the potential range of -500 to +1000 mV. Fifth cycles of voltammograms were given in figures, since electrochemical system approaches equilibrium by increasing the number of cycles.
Pyrite sample for FTIR spectroscopy was ground finely in an agate mortar, and then immediately transferred into polarization cell. Deoxygenated buffer solution was fed into completely sealed electrochemical cell in a closed system, and then polarization was applied for 10 min at -400, 200 and 800 mV. OVA-pyrite interaction was tested at a predetermined OVA dosage (500 mg/L) from preliminary experimental works. After polarization process, 2-3 mg pyrite sample was taken from cell, and ground in an agate mortar thoroughly with about 100 mg KBr. The ground mixture was then pressed under a pressure of 7 t for 5 min to produce highly- transparent 13 mm diameter KBr-disc pellet. IR spectra of pellets were recorded using Shimadzu 8300 FTIR spectrometer by averaging 30 scan at a resolution of 4 cm-1. Spectra were presented in transmittance mode.
Pyrite crystals were ground in a ceramic mill for flotation tests, and sized product in the range of 75-212 ��m was stored in glass tubes under N2 atmosphere to eliminate surface oxidation. Tests were made with 50 g sized sample in a cell having 750 ml effective volume (pulp solid rate is 6.67%) by a laboratory type sub- aerating Denver flotation machine. Impeller speed was adjusted to 1100 r/min. OVA dosage was applied as 50 g/t when required. Pyrite sample was polarized in flotation cell for 10 min at predetermined experimental conditions, and then, froth was skimmed for 5 min.
Flotation experiments were performed in a specially designed test set-up, as shown in Fig. 1. Surfaces in contact with flotation pulp were chosen from electrochemically inert materials to avoid undesired contamination of metal ions. The flotation cell was constructed from plexiglass, which had similar geometric shape with original cell supplied by producer. The reference and counter electrodes were mounted to each backside corners of cell, and protected with plexiglass curtains against adverse effect of turbulent condition keeping direct contact with pulp. The working electrode (about 1.5 m Pt wire) was fixed to the inner surface of cell so that efficient contact of mineral particles with Pt-wire was satisfied. Impeller, diffuser, and small part of mill which was in contact with pulp, were constructed from an engineering thermoplastic (derlin plastic�� polyoxymethylene) material. The plexiglass cell, derlin plastic mill with impeller and diffuser, and froth collecting pan were placed in a sealed transparent plastic box to satisfy oxygen-free environment. A thermoplastic rake was used to skim froth to the pan. Rake was mounted to the sealed box by rubber bellows enabling free movement of rake on froth zone. Similarly, mill, mounted to impeller, was set through rubber bellows and cover of sealed box. Rubber bellows were used to prevent inclusion of atmospheric O2 into closed cell system. N2 was blown continuously into the cup to provide oxygen-free environment within closed flotation cell system. Nitrogen was also used as flotation gas. Deoxygenated buffer solution was fed into cell in a closed system.
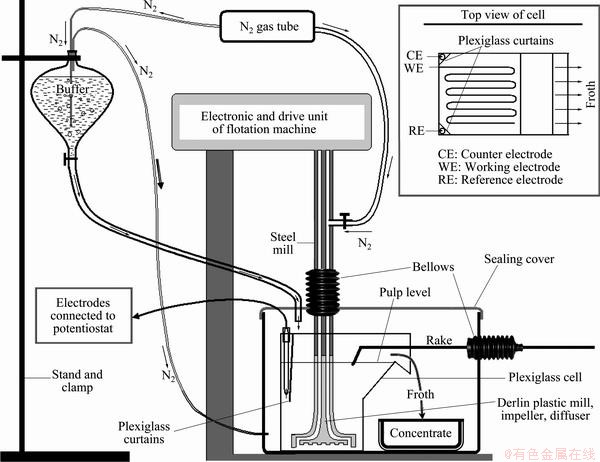
Fig. 1 Flotation set-up
3 Results and discussion
3.1 Role of ovalbumin on electrochemical behavior of pyrite
3.1.1 Electrochemistry of bare pyrite
Electrochemistry of pyrite was investigated as reference for subsequent works. Because electro- chemically active species (e.g. OVA) present in flotation pulp would alter redox behavior of pyrite, and determine types of surface products affecting floatability. Pyrite sample was polarized in reducing (-400 mV), slightly oxidizing (+200 mV) and highly oxidizing (+800 mV) conditions, and then FTIR spectra of polarized samples were drawn in the range of 4000-400 cm-1 (Fig. 2). OH- vibrations of hydrated Fe-hydroxides were obtained on the spectra between 4000-3000 cm-1, where water absorption bands did also appear [18,19]. Significant spectral differences were not obtained in the cited range depending on the applied potential. Then, interest was focused on the region of lower wave numbers where needed information exists. Strong absorption bands appeared at about 1630 cm-1, in the range of 1400-1350 cm-1 and around 1328 cm-1. The first one was referred to water absorption [19]. The third one around 1328 cm-1 was assigned to in-plane bending and symmetric vibration modes of species containing Fe-coordinated hydrolysis. The second one reaching its maxima at about 1380 cm-1 was assigned to OH in-plane bending vibrations of Fe-hydroxide. Pyrite oxidizes, and releases Fe2+ ions in oxidizing environment, which was followed by the precipitation of ferric species on mineral surface at alkaline pHs (reactions (1)-(5)). But, Fe2+ forms were main surface compounds at reducing potentials lower than -300 mV at pH of 9.2 [18]. Hydroxides of ferrous and ferric ions give closer IR peaks: Characteristic ferrous-hydroxide peak appeared at about 1370 cm-1 while it was about at 1380 cm-1 for ferric species. Then, ferrous-hydroxide band on pyrite spectrum of -400 mV could not clearly be discriminated due to the broad intense spectral formation at 1400-1350 cm-1.
FeS2 Fe2++2S+2e- (1)
Fe2++2S+2e- (1)
��=0.340+0.0296 lg[Fe2+]
Fe2++2H2OFe(OH)2+2H+ (2)
Fe2+Fe3++e- (3)
��=0.771+0.0592 lg([Fe3+]/[Fe2+])
Fe3++3H2OFe(OH)3+3H+ (4)
Fe(OH)2+OH-Fe(OH)3+e- (5)
��=-0.56-0.0592 lg[OH-]
The second product of pyrite oxidation was elemental sulfur (S). It oxidized to various sulfoxy species at metastable phases. Oxidation completes with the formation of stable sulfates at higher potentials on mineral surface in addition to metal-oxyhydroxides. In reducing conditions, however, S reduced to HS- in alkaline pulp [2,20]. Appearance of asymmetric stretching vibration peak of 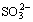 at about 1250 cm-1 on all the measured spectra in Fig. 2 proved the presence of metastable sulfoxy phases on pyrite. Characteristic bending (intense band around 600 cm-1), symmetric (weak concave shoulder at about 980 cm-1) and asymmetric (slightly intense peak at about 1094 cm-1 for 200 mV and 1109 cm-1 for 800 mV) S��O stretching vibrations of
at about 1250 cm-1 on all the measured spectra in Fig. 2 proved the presence of metastable sulfoxy phases on pyrite. Characteristic bending (intense band around 600 cm-1), symmetric (weak concave shoulder at about 980 cm-1) and asymmetric (slightly intense peak at about 1094 cm-1 for 200 mV and 1109 cm-1 for 800 mV) S��O stretching vibrations of 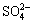 arose on pyrite spectra polarized in oxidizing (+200 mV, +800 mV) condition [21]. Small shift in peak point of IR band at about 1100 cm-1 was attributed to heavy surface coating with ferric- oxyhydroxides at highly oxidizing potentials rather than sulfate species. Intensities of sulfate related peaks were reasonably lower on pyrite spectrum polarized in reducing condition, even IR band at 1100 cm-1 disappeared.
arose on pyrite spectra polarized in oxidizing (+200 mV, +800 mV) condition [21]. Small shift in peak point of IR band at about 1100 cm-1 was attributed to heavy surface coating with ferric- oxyhydroxides at highly oxidizing potentials rather than sulfate species. Intensities of sulfate related peaks were reasonably lower on pyrite spectrum polarized in reducing condition, even IR band at 1100 cm-1 disappeared.
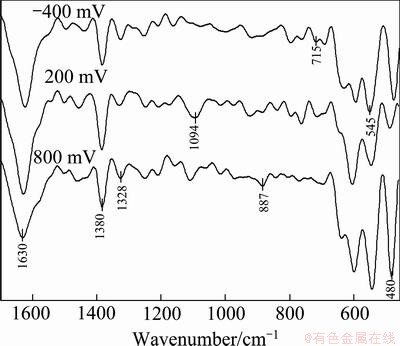
Fig. 2 FTIR spectra of pyrite polarized at different potentials
Discriminating peak structure was observed below 700 cm-1 along with slight variations in the range of 1000-700 cm-1: weak IR peaks at 916, 795, 760 and 715 cm-1, observed in reducing and slightly oxidizing conditions, disappeared on spectrum of pyrite polarized at 800 mV. Conspicuous variation in peak size was observed on primary intense bands at 480, 545, 596 and 633 cm-1 as well as weak band at 887 cm-1 at highly oxidizing potential. Such outcomes were mainly assigned to Fe-oxyhydroxides and sulfoxy species [21].
Potential scanning was applied using CV technique to clarify electrochemistry of pyrite besides polarization at certain potentials followed by FTIR spectroscopy analysis. Cyclic voltammograms were run in anodic direction reversing cathodic-lower scan limits from -250, -500, -600 and -750 mV, and fixing upper scan limit to +1000 mV (Fig. 3). Two anodic (A1, A2) and two cathodic (C1, C2) peaks were distinguished in scanned potential range. Peak A1 was attributed to Fe2+/Fe3+ redox couple [20]. This electrochemical process proceeded after pyrite oxidation releasing S0, which was not stable at oxidizing potentials. Therefore, sulfate was formed at higher scanning potentials. This oxidation process gave rise to a new anodic peak (A2) starting at about 740 mV. Such an intense peak could not be explained only with oxidation of S0. Since, the reaction rate of sulfate formation was slow, and the amount of sulfate formed depended on the amount of S0 released from pyrite surface [2,18,20]. In conformity with FTIR spectroscopy results, pyrite was thought to be covered with ferric-oxyhydroxides at higher potentials contributing to the intensity of peak A2. Additionally, water was not stable at higher potentials. Decomposition of water, and O2 evolution would also increase the size of A2.
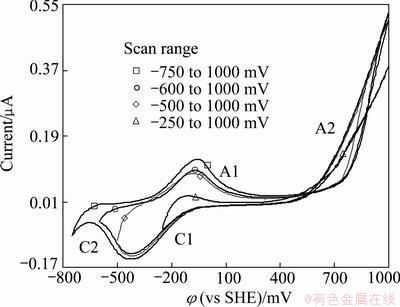
Fig. 3 Voltammograms fo pyrite measured at different scan ranges
In cathodic scan, peak C1 appeared in all the examined potential ranges. Peak A1 induced peak C1 in cathodic direction, and peaks A1 and C1 were accounted for Fe2+/Fe3+ redox couple as discussed in the foregoing paragraphs. Comparison of peak sizes of A1 and C1 affirmed that the number of electron released in anodic region for peak A1 was less than that for peak C1 in the cathodic sweep: QA1/QC1 values for scan ranges of -250 to 1000 mV, -500 to 1000 mV, -600 to 1000 mV and -750 to 1000 mV were 0.670, 0.684, 0.678 and 0.743, respectively. This situation indicated that some of the oxidized products at higher potentials (peak A2) would also be reduced to ferrous compounds or other related ones in cathodic scan, contributing to the strength of peak C1 [20]. It was interesting to note the significant rise on the rate of QA1/QC1 for the largest measured voltammogram range, which would be ascribed to variation in peak structure in cathodic scan: New reduction peak (C2) was obtained in cathodic sweep on the voltammogram measured in scanning range of -750 to 1000 mV. Peak C2 augmented the size of peak A1 as a result of re-oxidation of reduced product in pursuing anodic cycle. This redox process caused noticeable increase in the rate of QA1/QC1. This reduction peak was explained with the release of Fe (reaction (6)) [22].
Fe Fe2++2e- (6)
Fe2++2e- (6)
��=-0.44+0.0296 lg[Fe2+]
3.1.2 Electrochemistry of OVA-pyrite interaction
Proteins had net negative charge when solution pH was greater than isoelectric points (pI) of them (pI for OVA was 4.6 [13]). Similarly, pyrite had negative surface charge around open circuit potential-OCP at alkaline pHs (OCP was around 200 mV for pyrite at pH of 9.2 [2]). So, the amount of adsorbed OVA on pyrite via electrostatic mechanisms was expected to increase at positive scanning potentials above OCP due to supply of more binding sites. Further, OVA had electrochemically active sulfhydryl groups in addition to amine and carboxyl functional groups [11]. These groups were anticipated to manipulate OVA-pyrite interaction related with scanning potential. Therefore, pyrite voltammo- grams were also measured in the presence of OVA to elucidate the effect of potential on interaction of protein with mineral surface (Fig. 4). Potential scanning did not result in new peak as compared with pyrite voltammogram measured in the absence of OVA. Instead, surface passivation was observed in reducing to slightly oxidizing conditions: at first sharp and then gradual decrease in the intensity of peak A1 was seen depending on OVA concentration (Figs. 4 and 5), indicating the concentration dependence of OVA adsorption on pyrite surface. Current decay of peak A1 increased from 38.81% for 50 mg/L to 55.84% for 500 mg/L; but that of C1 was about at 35% for all OVA dosages. This would mean that even though OVA is still adsorbing on pyrite, its adsorption might be restricted in reducing conditions mainly due to the same charges of pyrite surface and OVA. Egg protein is known to be a ��hard�� protein, and had high internal stability. Therefore, OVA lost only a small fraction of its secondary structure (��-helix) during adsorption process on hydrophilic surface. It took a long time to reach equilibrium when conformational change or rearrangement of the structure of OVA occurred [23,24]. Then, its adsorption on pyrite in reducing environment was restricted. On the other hand, in slightly oxidizing environment, the degree of electron blocking action and the adsorption density of OVA became reasonable at higher bulk concentrations. The favored adsorption of OVA at slightly oxidizing potentials over negative conditions would chiefly be attributed to hydrophobic interaction [13,24] arising from interaction between OVA and hydrophobic S-containing redox products of pyrite. Pyrite under reducing condition was found to be oleophilic [25]. That is, even though it might not have measurable degree of hydrophobicity to satisfy floatability, it had certain degree of hydrophobic site for OVA adsorption. Hydrophobicity could increase especially with the formation of S0 around slightly oxidizing potentials by reaction (1).

Fig. 4 Voltammograms of pyrite measured at different OVA concentrations
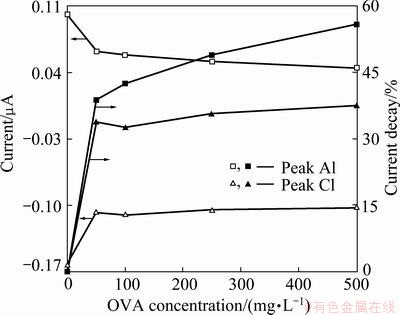
Fig. 5 Current change of peaks A1 and C1, and their current decay with OVA concentration
It is worth to mention here that OVA gave rise to about 100 mV shifts in the formation potential of peak A2, but any influence on strength could not be discriminated due to scan range. The rate of corrosion (decomposition of surface by electrochemical mechanisms) increased continuously until a certain potential was reached where passive layer formed on electrode surface and, corrosion rate decreased sharply. Corrosion of this passive layer restarted to proceed when potential applied was higher than the upper stability limit of passive region, where transpassive region was reached. In transpassive region, localized corrosion proceeded in weak passive sites, and the amount of electron transferred increased significantly with potential [2,17]. The locally corroded (oxidized) passive layer leading to peak A2 was estimated to be Fe-oxyhydroxides. As a result of further oxidation of pyrite surface, the reaction products interact electrochemically with physically adsorbed OVA to form metal-OVA chelate via sulfhydryl bonds of OVA [12,17]. The interaction of OVA with pyrite might also proceed through the formation of disulfide bond between sulfhydryl groups of OVA and sulfur containing (��SH) oxidation product of pyrite [10,16].
Interaction mechanisms of semiconducting sulfide minerals with flotation chemicals might not be clarified by voltammetry study at certain scan rates, and the characteristic peaks of adsorbed chemicals might not appear on voltammograms due to the kinetics of redox reactions expected to occur on the mineral surface. In some cases, lower scan rates and higher polarization times might be necessary for certain electrochemical processes, while the kinetics of electrochemical reactions might be fast enough to apply higher sweep rates in some other cases. Hence, the effect of scan rate on pyrite-OVA interaction was studied by CV technique at lower and higher sweep rates. The intensities of redox peaks increased proportionally at higher sweep rates. But, the appearance of new peaks or disappearance of present peaks was not observed on the measured voltammograms except a slight shift in the formation potential of peak A2.
FTIR spectroscopy study was also conducted to identify compounds formed on pyrite after polarization with OVA at predetermined potentials. OVA displayed simple and easily identifiable peak arrangement on reference spectrum despite its complex chemical structure (Fig. 6). The chemical constituent of OVA was found to be as follows: 50%-55% C, 6.9%-7.3% H, 15%-19% N, 0.3%-2.4% S and 19%-24% O [26]. So, the obtained strong FTIR peaks were mirrors of certain vibrations of bonds formed between major constituting elements. Diagnostic OVA peaks emerged strongly in the range of 1700-1500 cm-1: amide-I around 1655 cm-1, and amide-II at about 1530 cm-1. On the other hand, amide III vibrations caused two weak peaks at lower wavenumbers, 1306 cm-1 and 1240 cm-1. Native OVA shows a mixture of ��-sheet and ��-helix secondary structures about at the rates of 45% and 35%, respectively [23,27]. Bands assigned to ��-turn (1695-1660 cm-1), ��-helix (1660-1648 cm-1) and ��-sheet (1640-1620 cm-1) shaped amide-I band, and caused broad embodiment [19,28]. Sulfhydryl and disulfide groups of OVA caused weak vibrations below 700 cm-1 due to relatively lower rates of S (0.3%-2.4%) in the chemical composition of OVA. These bands mainly arose from disulfide bond and thiol groups [19,27,28].
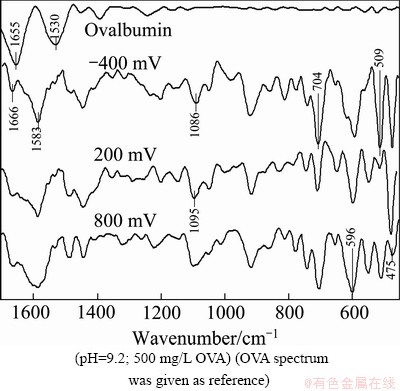
Fig. 6 FTIR spectra of pyrite polarized with OVA at -400 mV, 200 mV and 800 mV
The effect of applied polarization potential on electrochemical behavior of pyrite in the presence of OVA was discussed by measuring spectra, as shown in Fig. 6. Interaction made vibration bands harder to characterize due to structural changes. Major variations were observed both on the primary identification peaks of OVA (amide I and amide II in the range of 1700-1500 cm-1), and Fe-oxyhydroxide peak of pyrite at about 1380 cm-1. The appearance of new peaks and/or sharpened present/similar peaks between 1000-700 cm-1, and the overall decreasing trend in the intensity of absorption bands below 700 cm-1 were observed with the addition of OVA as compared with the bare pyrite spectra, as shown in Fig. 2. The rate of ��-helix decreased, while considerable increase of ��-sheet occurred due to the denaturation and interaction of OVA with mineral surface in alkaline condition. IR band of C=O groups of ��-turn at 1666 cm-1 became discernible as a result of change in the intensities of ��-helix vibrations [23,24,28]. Moreover, lower wavenumber ��-sheet vibrations in the range of 1640-1620 cm-1 could be distinguished in the same spectrum even though they were harder to be assigned due to the presence of broad sidechain COO- absorption at 1583 cm-1. These ��-sheet vibrations were referred to the hydration of amide C=O groups [27,28]. On the other hand, the disappearance of 1380 cm-1 and the emergence of various new peaks below 1200 cm-1 (e.g. 1045, 916, 738, 595 cm-1) would also assist the interaction and adsorption of OVA on pyrite in all the examined potentials.
OVA is known to be a hard protein. Then, its adsorption on pyrite in reducing condition is expected to be at lower rates because both pyrite and OVA have negative surface charges in aqueous solution at potentials lower than OCP in alkaline condition. Therefore, restricted conformational changes of OVA might occur due to its high internal stability. The disappearance (1380, 546 cm-1) and the appearance (1095, 1045, 810, 738, 704, 509 cm-1) of various IR peaks in reducing condition revealed the adsorption of OVA on pyrite (Fig. 6). Compared with the reference ones, these variations on pyrite-OVA spectrum of -400 mV would mainly be referred to the conformational changes of adsorbed OVA together with redox behavior of pyrite itself. It was evident that the denaturation of OVA occurred at reasonably lower rates in reducing condition, indicating the restricted amount of OVA adsorbed on pyrite due to the electrostatic repulsion between them. Therefore, variation on FTIR spectra would be explained with a large molecular size of OVA. Side-on-type configuration of denatured OVA perpendicular to the surface might explain the coverage of pyrite surface with lower amounts of protein. On the other hand, the intense absorption band at about 509 cm-1 should also be considered, where disulfide (S��S) vibration might form on FTIR spectrum of species containing S��S bond. Here, this band could not attribute to S��S stretching because disulfide stretching vibration was anticipated to appear with lower strength at the cited band [19]. Since sharp S��S band necessitated similar powerful C��S stretching vibration of disulfide at about 704 cm-1, which tended to give rise to very weak absorption unlike oxygen containing analogs at about 1095 cm-1 on the spectrum. In addition to cystine residue of OVA, disulfide bond could also form electrochemically through the interaction of cysteine residues, and/or by the reaction between OVA and SH- surface products of pyrite. These two mechanisms could only come true in oxidizing potentials [16,17]. Moreover, in-plane bending vibration of C��S��H should arise weakly at 916 cm-1 as compared with oxygen containing sulfur analogs. So, IR band of 509 cm-1 on pyrite-OVA spectrum for -400 mV (Fig. 6) would be referred to overlapped vibrations of other secondary structures formed as a result of denaturation, and OVA would be proposed to adsorb on pyrite electrostatically in reducing potential based on the evidences discussed.
Pyrite-OVA spectrum of 200 mV displayed similar band structure as compared with the spectrum of -400 mV, but at lower strengths beside depression of 509 cm-1 peak. This depression would be referred to hydrophobic interaction of OVA with pyrite, where the charges of both constituents were thought to have same sign. Hydrophobicity of pyrite surface increased in slightly oxidizing condition due to the formation of hydrophobic species, mainly S0, which led to the hydrophobic interaction between pyrite and OVA [24]. The presence of chemically active sites on mineral surface at OCP might be also evaluated as potential for the adsorption of OVA by chemical mechanisms [15]. Further increase in polarization potential from 200 mV to 800 mV resulted in a reasonable enhancement in the intensities of IR peaks almost as a whole. This increase was remarkable especially at 1583, 1095, 916, 704 and 509 cm-1, whereas conspicuous decrease at about 475 cm-1 occurred. Interesting variation at 509 cm-1 and 475 cm-1 would be explained with Fe-chelate formation [12]. Broadening of the band 596 cm-1 would assist this proposal.
Sulfate formation on pyrite occurred in oxidizing environment [20,21], which buought about asymmetric S��O stretching vibration around 1100 cm-1. Oxygen containing sulfur analogs of OVA might also contribute to this band. In neighboring region, C��H vibration of sidechains was also anticipated to form both in reducing and oxidizing conditions. Peak point of this band was at 1086 cm-1 at reducing potential, and shifted to 1095 cm-1 at oxidizing potentials with a slight increase in the intensity and broadness at higher potentials possibly due to the formation of ferric-sulfate species. Besides, slight deviation from 1109 cm-1 in the absence of OVA (Fig. 2) to 1095 cm-1 with OVA addition (Fig. 6) at highly oxidizing potential (800 mV) might show inhibition of ferric sulfate formation at a certain rate due to OVA adsorption.
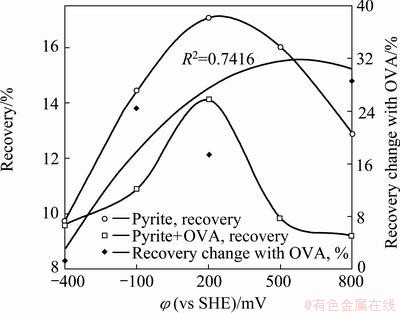
Fig. 7 Effect of polarization potential and OVA on floatability of pyrite
3.2 Role of ovalbumin on floatability of pyrite
The floatability of pyrite was investigated in collectorless condition in a wide potential range involving reducing (-400 mV, -100 mV), slightly oxidizing (200 mV �C around OCP) and highly oxidizing (500 mV, 800 mV) potentials in borate buffer solution (Fig. 7). Pyrite surface oxidized according to reaction (1), which started to proceed at 162.4 and 221.6 mV for Fe2+ concentrations of 10-6 and 10-4 mol/L, respectively [22]. Simultaneously, hydrophobic S0 and/or polysulfides released on mineral surface, which oxidized to sulfates at higher potentials. Ferrous component of pyrite oxidation process presented at metastable phase, and then oxidized to hydrophilic Fe3+-oxyhydroxides in oxidizing potentials at alkaline pHs (reactions (2)-(5)). These redox products manipulated pyrite floatability. In general, recovery data did not exceed 18% in the examined potential range. Recovery increased with potential up to about 200-250 mV, and then decreased slightly at higher potentials. Such low recovery values were explained with lower self-induced floatability of pyrite due to the presence of metal oxyhydroxides and/or sulfoxy compounds as leading hydrophilic surface species as discussed in the foregoing paragraphs [2,20,25].
Figure 7 shows the effect of OVA on the pyrite floatability. Depressing effect of OVA was in increasing order by potential. Recovery change was at minimal level in reducing conditions since both pyrite and natural form of OVA have negative surface charge. Although weak conformational changes of OVA molecule verify protein-pyrite interaction, electrostatic repulsion between them restricts the amount of adsorbed depressant [23,24]. OVA adsorption gave rise to an appreciable shrinkage on cathodic peak C1 (Fig. 4). The change in the intensity of peak C1 was found to be almost independent of bulk OVA concentration because the adsorption of denatured OVA was restricted to monolayer. Monolayer coverage was thought to be satisfied at extremely lower bulk OVA concentrations in reducing condition due to the large molecular size and side-on-type configuration [13]. Pyrite recovery was already at its minimum value in reducing condition, then such an electron blocking and depressing effect by electrostatic interaction could only be partially reflected on flotation results.
Hydrophobic species form on pyrite surface around slightly oxidizing potentials [2,20,22]. These species enhance the adsorption density of OVA due to hydrophobic interaction OVA and hydrophobic redox products present on pyrite. Then, the depressing effect of OVA increases gradually up to slightly oxidizing potential values although hydrophobic species might confine hydrophilic effect of OVA in slightly oxidizing conditions.
The rate of recovery drop with OVA reached its maxima in moderately oxidizing environment around 500 mV, where intense surface oxidation and formation of ferric species were expected on mineral surface. These compounds would favor the interaction of pyrite surface electrostatically with OVA molecules in addition to possible chemical interactions [4,9,15]. Moreover, hydrophobic S0 and/or polysulfides would oxidize to hydrophilic oxygen containing sulfur species, e.g. sulfates, by increasing the applied potential with considerably lower formation kinetics [2,20,22,25]. Synergetic effect of the adsorbed OVA and hydrophilic pyrite oxidation products caused remarkable depression in recovery especially in moderately oxidizing potential. On the other hand, the percentage decrease in pyrite recovery with OVA remained approximately unchanged at highly oxidizing potential, where the depression of pyrite with OVA would be attributed to the formation of complex OVA-metal chelates mainly by electrochemical mechanisms as proposed from CV and FTIR spectroscopy results. The role of interaction mechanisms on the floatability exhibited similar manner: percentage change in recovery was almost not noteworthy in oxidizing environment due to the absence of collecting agent in this work.
4 Conclusions
1) Pyrite floatability was low in the whole examined potential range in alkaline condition due to dominating hydrophilic Fe-compounds on pyrite surface in addition to sulfurous hydrophilic species. Hydrophobic elemental sulfur and/or polysulfides would form around slightly oxidizing condition, and improve the pyrite floatability.
2) The rate of pyrite depression by OVA drew parabolic curve in increasing order with potential, approaching equilibrium at mild to moderately oxidizing potentials.
3) Pyrite depression was at minimal level in reducing condition, which was attributed to weak conformational changes of OVA.
4) Effect of OVA became reasonable by increasing potential up to slightly oxidizing values, where hydrophobic interaction between OVA and hydrophobic S-containing species on pyrite was expected to occur despite dominating electrostatic repulsion.
5) At moderately oxidizing potentials over OCP of pyrite, OVA was thought to adsorb electrostatically in addition to chemical interactions. Depression of pyrite reached its maxima due presumably to the effective interaction between OVA and Fe-oxyhydroxides.
6) Electrochemical interaction was expected to occur at highly oxidizing potentials releasing hydrophilic OVA-metal chelates.
References
[1] SHEN W Z, FORNASIERO D, RALSTON J. Effect of collectors, conditioning pH and gases in the separation of sphalerite from pyrite [J]. Minerals Engineering, 1998, 11: 145-158.
[2]  T. A comparative evaluation of the response of platinum and mineral electrodes in sulfide mineral pulps [J]. International Journal of Mineral Processing, 2008, 87: 51-59.
T. A comparative evaluation of the response of platinum and mineral electrodes in sulfide mineral pulps [J]. International Journal of Mineral Processing, 2008, 87: 51-59.
[3] RATH R K, SUBRAMANIAN S, PRADEEP T. Surface chemical studies on pyrite in the presence of polysaccharide-based flotation depressants [J]. Journal of Colloid and Interface Science, 2000, 229: 82-91.
[4] BICAK O, EKMEKCI Z, BRADSHAW D J, HARRIS P J. Adsorption of guar gum and CMC on pyrite [J]. Minerals Engineering, 20: 996-1002.
[5] BOULTON A, FORNASIERO D, RALSTON J. Selective depression of pyrite with polyacrylamide polymers [J]. International Journal of Mineral Processing, 2001, 61: 13-22.
[6] PATRA P, NATARAJAN K A. Microbially induced flotation and flocculation of pyrite and sphalerite [J]. Colloids and Surfaces B: Biointerfaces, 2004, 36: 91-99.
[7] PATRA P, NATARAJAN K A. Role of mineral specific bacterial proteins in selective flocculation and flotation [J]. International Journal of Mineral Processing, 2008, 88: 53-58.
[8] POORTINGA A T, BOS R, NORDE W, BUSSCHER H J. Electric double layer interactions in bacterial adhesion to surfaces [J]. Surface Science Reports, 2002, 47: 1-32.
[9] JIA C, WEI D, LIU W, HAN C, GAO S, WANG Y. Selective adsorption of bacteria on sulfide minerals surface [J]. Transactions of Nonferrous Metals Society of China, 2008, 18: 1247-1252.
[10] CHEN J, LI Y, LONG Q. Molecular structure and activity of organic depressants for marmatite, jamesonite and pyrite flotation [J]. Transactions of Nonferrous Metals Society of China, 2010, 20: 1993-1999.
[11] FOTHERGILL L A, FOTHERGILL J E. Thiol and disulphide contents of hen ovalbumin. C-Terminal sequence and location of disulphide bond [J]. Biochemical Journal, 1970, 116(4): 555-561.
[12] STROHBEHN R E, FIGGINS J I. Method of separating components of technical eggs, edible eggs, yolk and whites and products therefrom: US 2008/0166447 A1 [P]. 2008.
[13] BASTRZYK A, POLOWCZYK I, SZELAG E, SADOWSKI Z. The effect of protein-surfactant interaction on magnesite rock flotation [J]. Physicochemical Problems of Mineral Processing, 2008, 42: 261-269.
[14] REZWAN K, MEIER L P, GAUCKPER L J. Lysozyme and bovine serum albumin adsorption on uncoated silica and AlOOH-coated silica particles: the influence of positively and negatively charged oxide surface coatings [J]. Biomaterials, 2005, 26(21): 4351-4357.
[15] LIU J, WANG Z, LI B, ZHANG Y. Interaction between pyrite and cysteine [J]. Transactions of Nonferrous Metals Society of China, 2006, 16: 943-946.
[16] ROJAS-CHAPANA J A, TRIBUTSCH H. Biochemistry of sulfur extraction in bio-corrosion of pyrite by Thiobacillus ferrooxidans [J]. Hydrometallurgy, 2001, 59: 291-300.
[17] BRETT C M A, BRETT A M O. Electrochemistry: Principles, methods, and applications [M]. Oxford: Oxford University Press, 1994: 427.
[18] CHERNYSHOVA I V. An in situ FTIR study of galena and pyrite oxidation in aqueous solution [J]. Journal of Electroanalytical Chemistry, 2003, 558: 83-98.
[19] BARTH A. Infrared spectroscopy of proteins [J]. BBA-Bioenergetics, 2007, 1767(9): 1073-1101.
[20]  H. Effect of galvanic interaction on collectorless flotation behaviour of chalcopyrite and pyrite [J]. international Journal of Mineral Processing, 1997, 52: 31-48.
H. Effect of galvanic interaction on collectorless flotation behaviour of chalcopyrite and pyrite [J]. international Journal of Mineral Processing, 1997, 52: 31-48.
[21] de DONATO P, KONGOLO M, BARRES O, YVON J,  F, BOUQUET E, ALNOT M, CASES J M. Chemical surface modifications of sulphide minerals after comminution [J]. Powder Technology, 1999, 105: 1-3, 141-148.
F, BOUQUET E, ALNOT M, CASES J M. Chemical surface modifications of sulphide minerals after comminution [J]. Powder Technology, 1999, 105: 1-3, 141-148.
[22] PETERS E. Thermodynamics and kinetic factors in the leaching of sulphide minerals from ore deposits and dumps [C]//SME-AIME Short Course on Bio Extractive Mining. Colorado, 1970: 46-75.
[23] HU H Y, DU H N. ��-to-�� structural transformation of ovalbumin: Heat and pH effects [J]. Journal of Protein Chemistry, 2000, 19: 177-183.
[24] NAKANISHI K, SAKIYAMA T, IMAMURA K. On the adsorption of proteins on solid surfaces, a common but very complicated phenomenon [J]. Journal of Bioscience and Bioengineering, 2001, 91: 233-244.
[25]  T. Two-liquid flotation of sulphides: An electrochemical approach [J]. Minerals Engineering, 2007, 20: 1246-1254.
T. Two-liquid flotation of sulphides: An electrochemical approach [J]. Minerals Engineering, 2007, 20: 1246-1254.
[26] EKMAN P,  O. Quantification of subnanomolar amounts of phosphate bound to seryl and threonyl residues in phosphoproteines using alkaline hydrolysis and malachite green [J]. Analytical Biochemistry, 1993, 214: 138-141.
O. Quantification of subnanomolar amounts of phosphate bound to seryl and threonyl residues in phosphoproteines using alkaline hydrolysis and malachite green [J]. Analytical Biochemistry, 1993, 214: 138-141.
[27] DONG A, MEYER J D, BROWN J L, MANNING M C, CARPENTER J F. Comparative Fourier transform infrared and circular dichroism spectroscopic analysis of ��1-proteinase inhibitor and ovalbumin in aqueous solution [J]. Archives of Biochemistry and Biophysics, 2000, 383: 148-155.
[28] JACKSON M, MANTSCH H H. The use and misuse of FTIR spectroscopy in the determination of protein structure [J]. Critical Reviews in Biochemistry and Molecular Biology, 1995, 30: 95-120.
Taki 1, Kemal 2, Sevil 3, 3
1. Department of Mining Engineering, Sitki University, 48170, Turkey;
2. Department of Metallurgical and Materials Engineering, Cumhuriyet University, Sivas 58140, Turkey;
3. Department of Mining Engineering, Cumhuriyet University, Sivas 58140, Turkey
ժ Ҫ�����嵰��(OVA)���׳Ƽ�������ף���һ�ֻ������������۵ı�����Լ������ķ��ӽṹ�к��е绯ѧ�����ϻ�������ѭ�����������������ѡʵ���о�OVA�ڻ����������������Ϊ������Ƽ����ڿ��ĵ�λ��Χ�ںͼ���pHֵ�����£��о��绯ѧ�������������ƺ������ʵ����á���OVA������ʱ���ɸ�������������λ�����´ﵽ����ֵ�������˵�λ��Χ���ɸ��Ի���������Fe�����������Լ���ˮ�����������͡����������Ե�λ��Χ��OVA-������������á�OVA������Ч�����ŵ�λ�ӻ�ԭ��λ����������λ����������Ŀ�·��λ�����ӣ�����Ҫ������OVA�������Ĺ���仯�Լ���·��λ�µ���ˮ��������ġ�OVA�����������е�������λ�´ﵽ����ֵ���ⱻ��Ϊ��������������OVA�Խ�����������ʽ�������ڽϸߵĵ�λ�����ָߵ�����Ч�����Լ���һֱ������ȥ��
�ؼ��ʣ���������ĭ��ѡ���Ѱ��ף�������ԭ��Ӧ������
(Edited by Chao WANG)
Foundation item: Project (M-279) supported by Cumhuriyet University Scientific Research Project Unit, Turkey
Corresponding author: Taki ; Tel: +90-252-2111654; Fax: +90-252-2111912; E-mail: takiguler@mu.edu.tr
DOI: 10.1016/S1003-6326(13)62795-8

