稀有金属 2012,36(01),74-79
Cr3+ 或V3+ 替代Mn3+ 对La0.4 Ca0.6 MnO3 电荷有序相的影响
彭振生 宋启祥 杨刚 王桂英 唐永刚 毛强
自旋电子与纳米材料安徽省重点实验室(培育基地)
中国科学技术大学合肥微尺度物质科学国家实验室
宿州学院电子与电气工程系
摘 要:
用固相反应法制备了La0.4Ca0.6Mn1-xCrxO3(LCMCO)和La0.4Ca0.6Mn1-yVyO3(LCMVO)(x,y=0.00,006,0.08)多晶样品。通过XRD、M-T曲线、ESR谱线,研究了Cr3+或V3+替代Mn3+对La0.4Ca0.6MnO3电荷有序相的影响。实验结果表明:电荷有序相随着Cr掺杂浓度的增加而被破坏,在LCMCO体系中电荷有序相几乎完全消失;而当V掺杂时,虽然电荷有序相随着V成分的增加会逐渐变弱,但电荷有序相依然存在于LCMVO体系中。用V3+替代Mn3+只是对于长程的电荷有序仅仅起了隔断的作用;用Cr3+替代Mn3+破坏了CE型反铁磁的自旋序从而引起电荷序的融化。从实验上证明了电荷序CE型反铁磁体系中,电荷序和自旋序存在强耦合相互作用。
关键词:
钙钛矿锰氧化物 ;电荷有序 ;V掺杂 ;Cr掺杂 ;
中图分类号: O482.54
作者简介: 彭振生(1948-),男,安徽怀远人,教授;研究方向:凝聚态物理(E-mail:ahpengzhsh1948@126.com);
收稿日期: 2011-02-10
基金: 国家自然科学基金项目(19934003); 安徽省教育厅自然科学研究项目(KJ2011A259,KJ2009A053Z);安徽省教育厅自然科学研究项目(KJ2009B281Z,KJ2010B228,KJ2010B229)资助;
Influence of Low Cr3+ or V3+ Substitute Mn3+ on Charge Ordering Phase in La0.4 Ca0.6 MnO3
Abstract:
Polycrystalline samples of La0.4Ca0.6Mn1-yCryO3(LCMCO) and La0.4Ca0.6Mn1-xVxO3(LCMVO)(x,y=0.00,0.06,0.08) were prepared by the solid state reaction method.The influence of Cr3+ or V3+ substitution for Mn3+ on the magnetic property and charge ordering phase of La0.4Ca0.6MnO3 were studied through the measurements of X-ray diffraction(XRD),magnetization-temperature(M-T) curves and ESR spectra.With the increase of the amount of Cr,the charge ordering phase of La0.4Ca0.6MnO3 was destroyed,and the charge ordering phase in the LCMCO system disappeared almost.On the other hand,when the V was doped,the charge ordering phase was gradually weakened with the increase of V.However,the charge ordering phase still existed in the LCMVO system.That was because that the V3+ substitution for Mn3+ only played a role in the partition of the long-range charge ordering.In addition,the Cr3+ substitution for Mn3+ destroyed the spin order of CE-type antiferromagnetism,which led to the melting of charge ordering.The results confirmed that a strong coupling interaction between charge order and spin order existed in the charge order CE-type antiferromagnetic system.
Keyword:
perovskite manganite;charge ordering;V doping;Cr doping;
Received: 2011-02-10
在过去的十多年里,钙钛矿氧化物因为其有趣的基本结构,电磁特性及潜在的在磁感应和磁存储方面的应用,受到了很多关注
[1 ,2 ,3 ,4 ]
。在RE1-x Tx 3 体系中空穴型掺杂已经被人们进行了详细的研究(RE为三价稀土离子,T为半径较大的正一价或者正二价的阳离子)。为了更好的了解这些氧化物的内在机制,目前很多工作都集中于对电荷有序行为的研究
[5 ,6 ]
。电荷有序通常被认为是混价的Mn3+ 和 Mn4+ 离子在实空间的周期性排列,它往往伴随着轨道有序和反铁磁(AFM)转变的发生。一般说来,电荷有序(change ordering, CO)相起源于库仑势,轨道有序,Jahn-Teller效应。实验发现,磁场、电场、电磁辐照等手段都能破坏电荷有序或者削弱其强度
[7 ,8 ,9 ]
。对于那些具有较小A位离子半径的电荷有序(CO)锰氧化物,发现即使在高达27 K的磁场下都难以破坏CO相
[10 ]
。相比之下,通过Al 3+ ,Ga3+ ,W6+ 等对Mn3+ /Mn4+ 进行元素替代,可以有效地破坏CO相的稳定性
[11 ,12 ,13 ,14 ]
。
选用的母体材料La0.4 Ca0.6 MnO3 是CE型电荷有序反铁磁材料
[15 ]
,用磁性离子Cr3+ 或非磁性离子V3+ 替代Mn3+ ,本文通过对比来研究体系中的电荷序和自旋序之间的耦合相互作用。
1 实 验
采用标准固相反应法制备La0.4 Ca0.6 Mn1- x x 3 (LCMCO)和La0.4 Ca0.6 Mn1- y y 3 (LCMVO)(x ,y =0.00,006,0.08)多晶样品。制备过程如下:将高纯度的La2 O3 在600 ℃下脱水6 h(因为La2 O3 极易吸潮),与高纯度的CaCO3 ,CrCO3 ,V2 O3 ,MnO2 试剂按化学计量进行配比,充分混合并研磨后,在900 ℃预烧12 h,自然冷却后,取出样品仔细研磨,在1100 ℃锻烧12 h。重复上述过程,再在1300 ℃锻烧12 h,以获得良好的结晶。将样品压成直径为13 mm,厚度约为1 mm 的圆片,在1380 ℃烧结24 h,最后切割成长条状样品。样品的晶体结构分析在日本玛珂公司18 kW粉末X射线衍射仪(MXP18AHF)上进行,采用Cu靶Kα射线(λ=0.1542 nm)。M -T 曲线的测量使用的是超导量子干涉仪(SQUID),分别在0 T和0.01 T磁场中将样品冷却到5 K,再升温测量。电子自旋共振谱(ESR)的测量在ER-200D共振谱仪上进行,使用的是粉末样品,微波频率为9.61 GHz,测量谱为微分吸收谱,测量温区为100~300 K。
两组样品La0.4 Ca0.6 Mn1- x x 3 (LCMCO)和La0.4 Ca0.6 Mn1- y y 3 (LCMVO)(x ,y =0.00, 006, 0.08),用磁测量,电子自旋共振的方法进行分析研究。V掺杂是用来与Cr掺杂做参照物,原因如下:(1)Cr3+ 和V3+ 离子取代Mn3+ 离子时都表现出+3价,它们都是非Jahn-Teller活性离子,而且半径相近(Cr3+ 半径为0.062 nm,V3+ 半径为0.064 nm,Mn3+ 半径0.064 nm),替代后由晶格不匹配引起的晶格畸变很小,可以忽略不计;(2)因为V3+ 表现为非磁性离子,而Cr3+ 是磁性离子,它的电子结构与Mn4+ 相同(t 2g 3 e g 0 ),Cr3+ 离子替代Mn3+ 离子可以破坏自旋序从而引起电荷序的破坏。研究结果表明:当少量的Cr3+ 离子替代Mn3+ 离子后就可以在反铁磁绝缘体中抑制电荷有序,并且建立了一些铁磁金属团簇。
2 结果与讨论
利用X射线衍射对La0.4 Ca0.6 Mn1- x x 3 (LCMCO)和La0.4 Ca0.6 Mn1- y y 3 (LCMVO) (x ,y =0.00,006,0.08)多晶样品的晶体结构进行检测,从XRD测量结果表明,所有样品都具有单相正交钙钛矿结构,没有杂相出现。
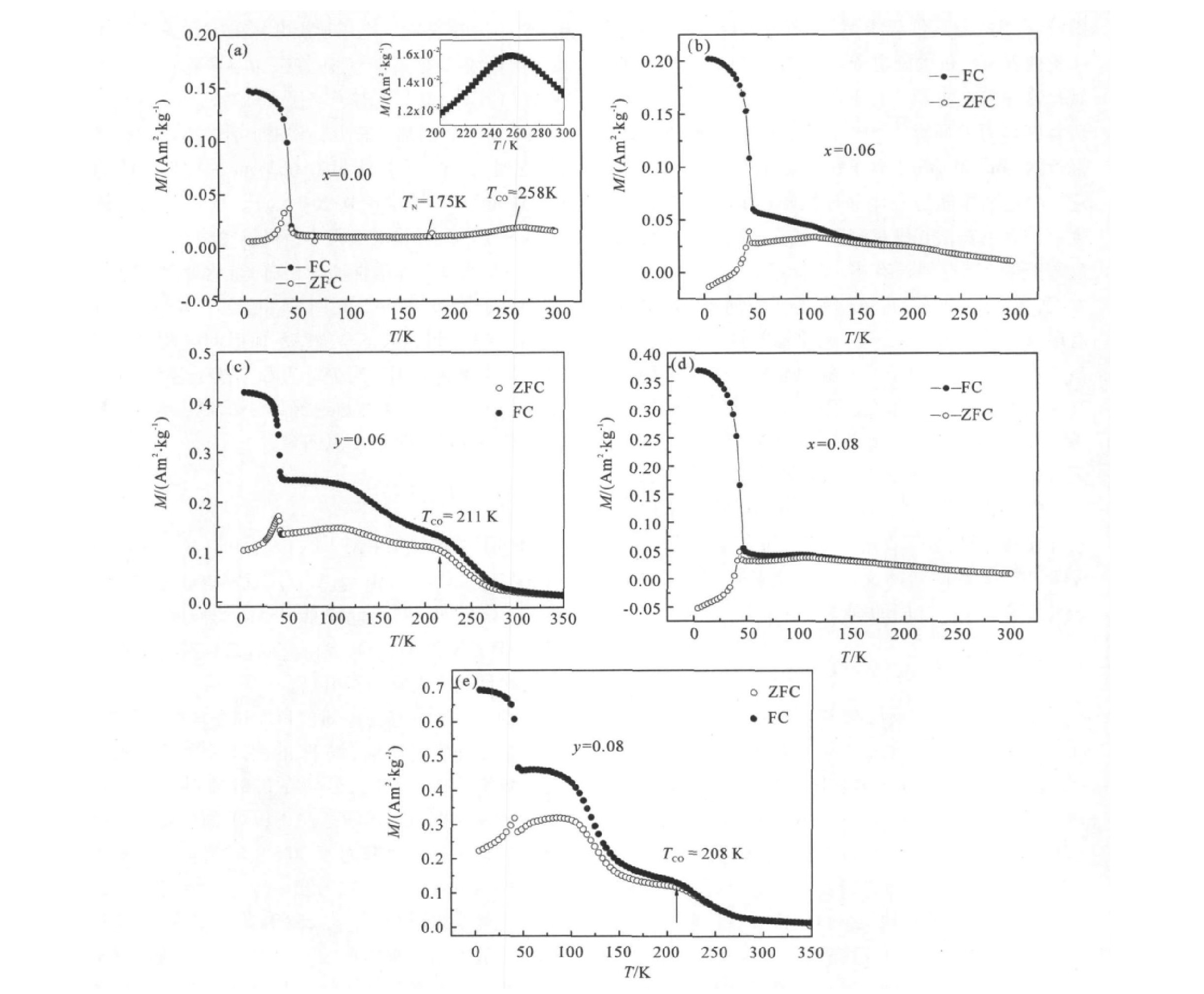
图1表示在零场冷却(ZFC)和加场冷却(FC)下的磁化强度对温度(M -T )的曲线。样品首先在无磁场下冷却到5 K,然后在0.01 T的外磁场下升温到300 K。FC曲线是在0.01 T的外磁场下降温到5 K之后,再升温至300 K时测量的。对于母体样品,从图1可以看出:随温度降低,样品的磁化强度逐渐增大,在258 K附近出现峰值(见插图中放大了的M -T 曲线),在258 K出现的峰值对应于电荷有序温度T CO ,此时出现CE型反铁磁有序相
[16 ]
;在电荷有序转变温度以下直到175 K,磁化强度逐渐缓慢减小,意味着样品中少量顺磁(PM)向反铁磁(AFM)转变;从175 K到50 K磁化强度基本不变,这意味着长程的CO-AFM相发生了,175 K对应的是Néel温度,即T N =175 K;当温度降到约41 K时,零场冷却曲线的磁化强度M 又迅速升到最大值,出现一个明显尖峰,温度低于41 K时磁化强度随温度降低又迅速下降,这是自旋玻璃态的特征
[17 ]
。
在Cr掺杂样品La0.4 Ca0.6 Mn1- x x 3 (LCMCO)中,如图1(b,d)所示,对于x =0.06的样品,随温度降低M 值持续增大,意味着电荷有序相已经基本坍塌;温度降至110 K左右出现鼓包,110 K对应的是Néel温度,即T N =110 K。在 200 K以下,ZFC对应的M -T 曲线与FC对应的M -T 曲线发生明显分叉,说明样品在反铁磁背景下有少量铁磁成分。温度降至41 K左右时零场对应的M -T 曲线仍出现尖峰,说明仍保持自旋玻璃态特征;对于x =0.08的样品,随温度降低M 值持续增大,意味着电荷有序相已被完全融化;温度降至110 K左右时M 出现一个峰值,这个峰对应的温度是Néel温度,即T N =110 K;温度降至41 K左右时仍保持自旋玻璃态特征;在110 K以下,零场与加场对应的M -T 曲线发生分离,说明样品在反铁磁背景下有少量铁磁成分,产生了相分离。
图1 La0.4Ca0.6Mn1-xCrxO3和La0.4Ca0.6Mn1-yVyO3系列样品的M-T曲线
Fig.1 M -T curves of La0.4 Ca0.6 Mn1-x Crx 3 and La0.4 Ca0.6 Mn1-y Vy 3 samples
在V掺杂样品La0.4 Ca0.6 Mn1- y y 3 (LCMVO)中,如图1(c,e)所示,对于y =0.06和y =0.08的两个样品,电荷有序相仍得到保持,电荷有序温度T co 分别降至211及208 K。 当温度降到40 K左右时,ZFC和FC对应的M -T 曲线出现分叉现象,ZFC曲线出现尖峰,呈现自旋玻璃态的典型特征。ZFC和FC对应的M -T 曲线产生分叉现象,说明样品在反铁磁背景下有少量铁磁成分。
从M -T 曲线上来看,V掺杂和Cr掺杂存在着很大的差别,虽然掺杂量是一样的,但是随着V掺杂量增大,T CO 被压制到比较低的温度,电荷有序相仍得以保持,而随着Cr的掺杂,却没有发现任何有关电荷有序相形成的信号。
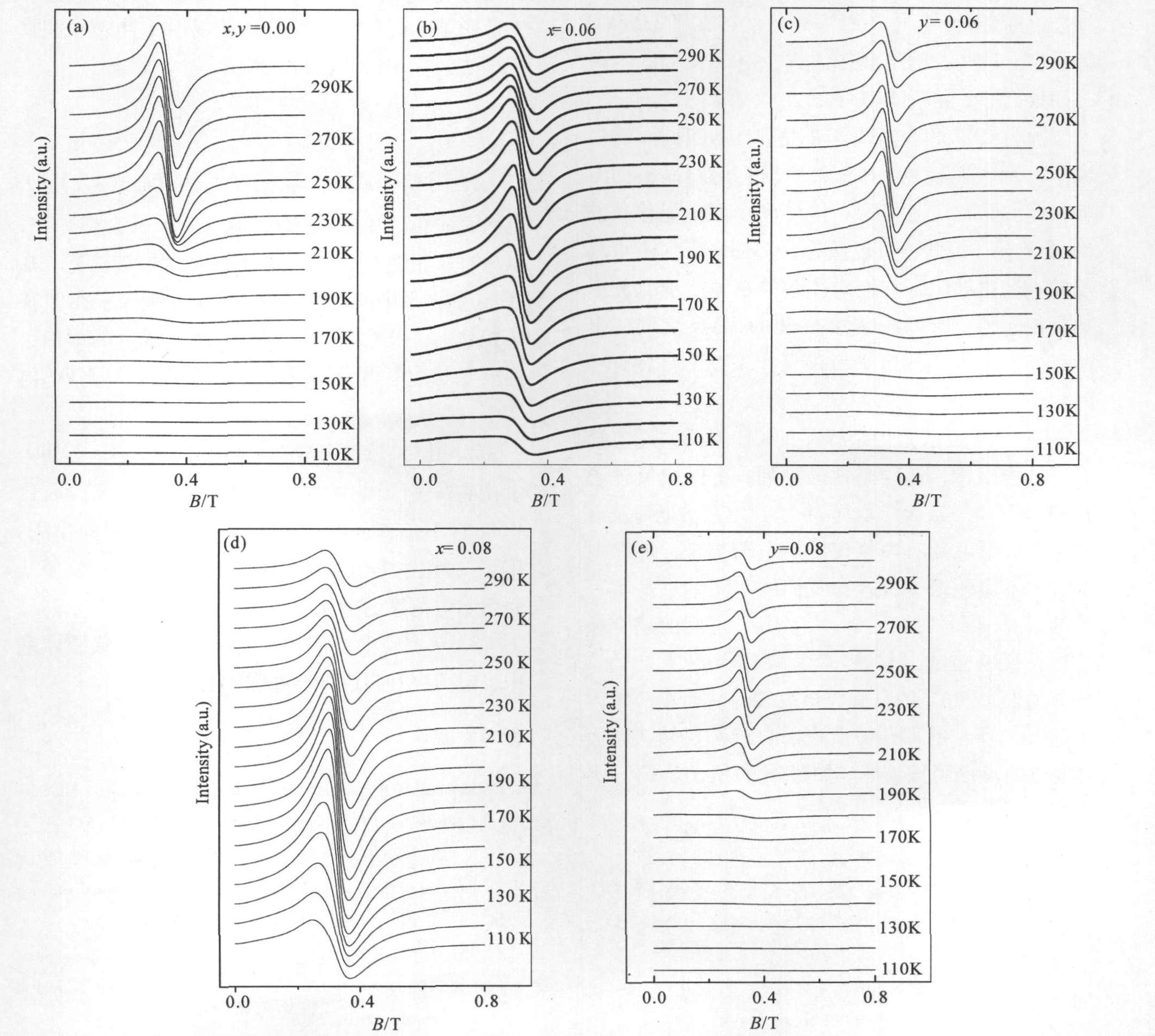
利用电子自旋共振(ESR)的方法对体系的微观磁性作了研究, 结果如图2所示。
对于母体样品,在温度高于T co (258 K)时,所有的ESR信号都有一个洛伦兹线谱吸收峰,其朗德因子g ≈2.0,这个g ≈2.0信号被认为是顺磁吸收峰。在温度低于T CO 时,随温度降低峰强逐渐减小,且峰位向低场移动,说明T CO 以下随温度降低顺磁成分减少,产生少量铁磁成分。在T co以下没有观察到反铁磁信号,说明反铁磁耦合很强。
在Cr掺杂样品La0.4 Ca0.6 Mn1-x Crx 3 (LCMCO)中,如图2(b,d)所示,两个样品在测量温度范围内,所有的ESR信号都有一个洛伦兹线谱吸收峰,随温度降低峰强增大峰位向低场移动,说明电荷有序相已被融化,铁磁成分增多。
在V掺杂样品La0.4 Ca0.6 Mn1-y Vy 3 (LCMVO)中,如图2(c,e)所示,对于y =0.06和y =0.08的两个样品,在210 K以上(居里温度T CO 分别为211和208 K)所有的ESR信号都有一个洛伦兹线谱吸收峰,其朗德因子g ≈2.0。在居里温度T CO 以下,随温度降低峰强减小,保持母体的ESR谱特征,说明电荷有序相仍得到保持。
图2 La0.4Ca0.6Mn1-xCrxO3和La0.4Ca0.6Mn1-xVxO3的ESR谱
Fig.2 ESR spectra of La0.4 Ca0.6 Mn1-x Crx 3 and La0.4 Ca0.6 Mn1-y Vy 3 samples
通过对5个样品的ESR谱分析可以看出,5个样品的微观磁结构与宏观磁性相一致。
如上讨论,电荷有序相随着Cr掺杂浓度的增加而被破坏,在LCMCO体系中电荷有序相几乎完全消失。因此,Cr掺杂导致了电荷有序相的融化;而当V掺杂时,虽然电荷有序相随着V成分的增加会逐渐变弱,但电荷有序相依然存在于LCMVO体系中。这些表明虽然掺入V,对于长程的电荷有序仅仅起了隔断的作用。
因为Cr3+ 和V3+ 离子都是非Jahn-Teller活性离子,而且两者半径相似,所以它们对晶格的影响相同。同时,巡游的eg 传导电子都不能移动到Cr或V位。因此,可以认为它们都表现为不可移动的eg 轨道缺陷,它们对体系的轨道序变化的影响是相同的。所以,LCMCO和LCMVO之间的电荷有序发生变化的区别应该只能来源于自旋有序的变化。
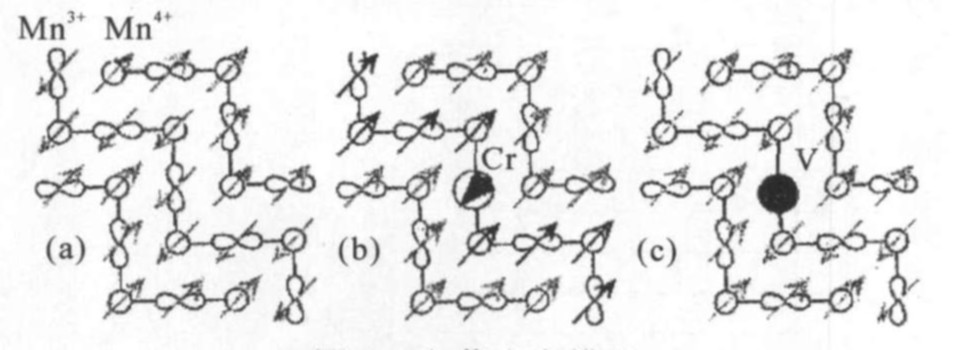
图3(a)表示未掺杂样品的CE型反铁磁电荷有序结构模型,链内呈铁磁排列,链间呈反铁磁排列。当Cr3+ 或V3+ 取代Mn3+ ,体系的电荷没有变化。如果电荷有序仅仅是由电荷决定,那么电荷有序不应该改变,这与我们的实验结果是不一致的。对于Cr3+ 掺杂,Cr3+ ~Mn4+ 之间的超交换作用是反铁磁的
[18 ]
,当Cr3+ 取代Mn3+ 后,Cr3+ 相邻的两个Mn4+ 的自旋应该反转,从而小区域内引起Cr3+ 所在的那条链所有的自旋都反转,因而体系的自旋序将被破坏。如图3(b)所示,Cr3+ 所在的那条链和它相邻的两条链就形成了铁磁排列,导致铁磁区域变大,这样会增强Mn3+ 上的e g 电子的巡游能力,压制体系中的电荷有序效应。随着越来越多的Cr3+ 替代Mn3+ ,铁磁区域变的越来越大,e g 电子的巡游能力也变得更强,从而最终破坏了电荷有序,因此自旋序的破坏将会引起电荷序难以形成。对于V掺杂,如图3(c)所示,V3+ 离子是非磁性离子,当V3+ 取代Mn3+ 时,V3+ 离子可以看为“自旋空穴” 。这里,“自旋空穴”仅仅把体系中长程的自旋序破坏为短程的自旋序,因此电荷有序在所有的样品中都能发现。这从实验上证明了电荷序CE型反铁磁体系中,电荷序和自旋序之间存在强耦合相互作用。
图3 电荷有序模型
Fig.3 Model of charge order
(a)Maternal CE-type AFM order model;(b)Magnetic Cr3+ ions substitution for Mn3+ ;(c)Non-magnetic V3+ ions substitution for Mn3+
3 结 论
1.用Cr3+ 或V3+ 取代Mn3+ 对La0.4 Ca0.6 MnO3 电荷有序相的影响大不相同。在Cr掺杂样品中,电荷有序相随着Cr掺杂浓度的增加而被破坏,在LCMCO体系中电荷有序相几乎完全消失;在V掺杂样品中,虽然电荷有序相随着V成分的增加会逐渐变弱,但电荷有序相依然存在于LCMVO体系中。
2.用Cr3+ 或V3+ 取代Mn3+ 对La0.4 Ca0.6 MnO3 电荷有序相影响的物理机制不同。用V3+ 取代Mn3+ 对于长程的电荷有序仅仅起了隔断的作用;用Cr3+ 替代Mn3+ 破坏了CE型反铁磁的自旋序从而引起电荷序的融化。
3.从实验上证明了电荷序CE型反铁磁体系中,电荷序和自旋序存在强耦合相互作用。
参考文献
[1] Salamon M B,Jaime M.The physics of manganites:structureand transport[J].Rev.Mod.Phys.,2001,73(3):583.
[2] Rao C N R,Arulraj A,Cheetham A K,Bareau B.Chargeordering in the rare earth manganates:the experimental situation[J].J.Phys:Condens.Matter.,2000,12(7):R83.
[3] Mao Q,Guo H Y,Wang G Y,Peng Z S.Electric-magnetic prop-erties of La0.7-xDyxSr0.3CoO3 system[J].Chinese Journal ofRare Metals,2010,34(5):705.(毛强,郭焕银,王桂英,彭振生.La0.7-xDyxSr0.3CoO3体系磁电性质研究[J].稀有金属,2010,34(5):705.)
[4] Moritomo Y,Asamitsu A,Kuwahara H,Tokura Y.Giant mag-netoresistance of manganese oxides with a layered perovskitestructure[J].Nature,1996,380(1):141.
[5] Wang G Y,Yan G Q,Peng Z S,Liu N.Influence of doping Von transport properties and charge ordering of La0.45Ca0.55MnO3[J].Chinese Journal of Rare Metals,2010,34(3):345.(王桂英,严国清,彭振生,刘宁.V掺杂对La0.45Ca0.55MnO3电荷有序态及输运性质影响的研究[J].稀有金属,2010,34(3):345.)
[6] Zhang M Y,Peng Z S,Guo H Y,Mao Q,Yan G Q.Destruc-tion of charge-ordering phase and Influence on magntic propertyinduced by W doping at Mn site of La0.3Ca0.7MnO3[J].Journalof Rare Earths,2006,24(S2):192.
[7] Fiebig M,Miyano K,Tomioka Y,Tokura Y.Visualization ofthe local insulator-metal transition in Pr0.7Ca0.3MnO3[J].Sci-ence,1998,280(537):1925.
[8] Oshima H,Miyano K,Konishi Y.Kawasaki M,Tokura Y.Switching behavior of epitaxial perovskite manganite thin films[J].Appl.phys.lett.,1999,75(10):1473.
[9] Kiryukhin V,Kim B G,Katsufuji T,Hill J P,Cheong S W.Nanoscale anisotropic structural correlations in the paramagneticand ferromagnetic phases of Nd0.5 Sr0.5 MnO3[J].Phys.Rev.B.,2001,63(14):144406.
[10] Tokunaga M,Miura N,Tomioka Y,Tokura Y.High-magnetic-field study of the phase transitions of R1-xCaxMnO3(R=Pr,Nd)[J].Phys.Rev.B.,1998,57(9):5259.
[11] Li R W,Sun J R,Wang Z H,Zhang S Y,Tang N S,Bao G.Effects of the relative proportion of ferromagnetic and charge-or-dered phases on the metal-insulator transition temperature in La0.5Ca0.5 Mn1-x Gex O3[J].J.Appl.Phys.,2000,88(12):7041.
[12] Morikawa O,Tonouchi M,Hangyo M.Sub-THz spectroscopic sys-tem using a multimode laser diode and photoconductive antenna[J].Appl.Phys.Lett.,1999,75(24):3772.
[13] Guo H Y,Zhang S X,Yan G Q.Establishment and melting ofcharge ordering in CaMn1-xWxO3(0.05≤x≤0.20)system[J].Chinese Journal of Rare Metels,2006,30(1):6.(郭焕银,张世雄,严国清.CaMn1-xWxO3体系电荷有序的建立与消失[J].稀有金属,2006,30(1):6.)
[14] Peng Z S,Guo H Y,Yan G Q,Mao Q.Magnetic properties ofperovskite manganite La0.3Ca0.7Mn0.96W0.04O3 in charge orderingphase[J].Chinese Journal of Rare Metels,2006,30(5):635.(彭振生,郭焕银,严国清,毛强.钙钛矿锰氧化物La0.3Ca0.7Mn0.96W0.04O3在电荷有序相中的磁性质[J].稀有金属,2006,30(5):635.
[15] Pissas M,Kallias G.Phase diagram of the La1-xCaxMnO3 com-pound(0.5<x<0.9)[J]Phys.Rev.B,2003,68(13):134414.
[16] Tomioka Y,Okuda T,Okimoto Y,Asamitsu A,Kuwahara H,Tokura Y.Charge/orbital ordering in perovskite manganites[J].Journal of Alloys and Compounds,2001,326(1-2):27.
[17] Liang Y,Peng Z S,Yan G Q,Guo H Y,Cai Z R.Magneticproperties of perovskite oxide La0.3Ca0.7MnO3[J].Chinese Jour-nal of Rare Metals,2005,28(4):513.(梁燕,彭振生,严国清,郭焕银,蔡之让.钙钛矿氧化物La0.3Ca0.7MnO3的磁性质[J].稀有金属,2005,28(4):513.)
[18] Goodenough John B.Magnetism and the Chemical Bond[M].New York:Interscience,1963.