Trans. Nonferrous Met. Soc. China 23(2013) 3400-3404
First principles calculation of electronic structure, chemical bonding and elastic properties of ultra-incompressible Re2P
Yi-fu WANG1, Qing-lin XIA2, Yan YU2
1. Key Laboratory of High Performance Computing and Stochastic Information Processing (Ministry of Education of China), College of Mathematics and Computer Science, Hunan Normal University, Changsha 410081, China;
2. School of Physics and Electronics, Central South University, Changsha 410083, China
Received 22 January 2013; accepted 18 September 2013
Abstract: The electronic structures, chemical bonding and elastic properties of the Co2P-type structure phase ultra-incompressible Re2P (orthorhombic phase) were investigated by density-functional theory (DFT) within generalized gradient approximation (GGA). The calculated energy band structures show that the orthorhombic structure phase Re2P is metallic material. The density of state (DOS) and the partial density of state (PDOS) calculations show that the DOS near the Fermi level is mainly from the Re-5d state. Population analysis suggests that the chemical bonding in Re2P has predominantly covalent character with mixed covalent-ionic character. Basic physical properties, such as lattice constant, bulk modulus, shear modulus, and elastic constants Cij, were calculated. The elastic modulus and Poisson ratio were also predicted. The results show that the Co2P-type structure phase Re2P is mechanically stable and behaves in a brittle manner.
Key words: Re2P; first principles; electronic structures; chemical bonding; elastic properties
1 Introduction
The searching for new superhard and ultra- incompressible materials with prominent physical and chemical properties is one of the most attractive research fields [1-6]. Transition-metal (TM) borides, carbides and nitrides are of great interest and importance for their unique properties, such as good chemical stability, high strength, high hardness, high ultra-incompressibility, high melting point, low electrical resistivity and high thermal conductivity [7-13]. These properties are ascribed to the directional bonding in covalent networks of atoms coordinated by a small number of ligands with short interatomic distances, which consist of atoms with low Z elements, such as Be, B, C, N, O, Al, Si, or P.
However, there are few reports on the physical properties of hardness and incompressibility of materials with slightly heavier low Z elements (e.g., Si, P or S) in detail because the values for hardness and incompressibility are thought to be lower than the expected ones compared with the incorporation of B or N [14]. Recently, in situ high-pressure experiments and theoretical calculations have classified Co2P-type structures phase Re2P into the ultra-incompressible materials with a bulk modulus of above 300 GPa [14]. Despite the experimental and theoretical researches on Re2P, few is known regarding the relationship of their chemical bonding and mechanical properties. Motivated by these observations, in this work, a systematic first principles study on the electronic structure, chemical bonding and elastic properties of the orthorhombic phase Re2P.
2 Calculation details
The first principles calculations described here are based on DFT using a plan-wave expansion of the wave function [15,16]. The exchange correlation energy is calculated by the GGA with the Perdew-Burke- Ernzerhof (PBE) functionality [17]. The ionic cores are represented by ultra-soft pseudopotentials for Re and P atoms. The Re 5s25p65d56s2 electrons and P 3s23p3 electrons are explicitly treated as valence electrons. The Monkhorst and Pack scheme of k-point sampling is used for integration over the first Brillouin zone [18]. The cutoff energy is chose to be 500 eV, and the Brillouin- zone sampling k-point set mesh parameters are 6��8��3. The set of parameters assure the total energy convergence of 5.0��10-6 eV/atom, the maximum force of 0.01 eV/ 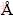 , the maximum stress of 0.02 GPa and the maximum displacement of 5.0��10-4 .
, the maximum stress of 0.02 GPa and the maximum displacement of 5.0��10-4 .
3 Results and discussion
3.1 Geometry and structure properties
The crystal structure of the Co2P-type structure phase Re2P belongs to the space group Pnma (No. 62, Z=4), which puckers Re-Re and Re-P strands in the [100] direction, straightens Re-Re-P stacking in the [001] direction and linear strands of Re and P atoms in the [010] direction as depicted in Fig. 1. There are inequivalent atomic positions: Re 1 at 4c site (0.8426, 1/4, 0.0744), Re 2 at 4c site (0.8462, 1/4, 0.7828), and P at 4c site (0.356, 1/4, 0.1425) [14]. The experimental lattice parameters are a=5.5464 , b= 2.9421 and c=10.0483 .

Fig. 1 Crystal structure of Re2P
At the first stage, the full structural optimization of this phase is performed both over the lattice parameters and the atomic positions including the internal coordinate. The calculated optimization lattice parameters a, b, c, V and atomic coordinates compared with available experimental data [14] for Re2P are summarized in Table 1, which shows that the calculated values by GGA are in agreement with the available experimental and theoretic data.
3.2 Electronic and chemical bonding
The calculated energy band structure of Re2P along with the high-symmetry points of the Brillouin-zone by GGA shows that the energy band curves pass through Fermi energy (EF) level (Fig. 2(a)), indicating that Re2P is metallic compound. The total densities of states (TDOS) and partial densities of states (PDOS) for Re2P are plotted in Fig. 2(b). The DOS at the Fermi level (EF) locates at the bottom of a valley and originates mainly from the Re-5d electrons.
The Mulliken bond population is useful for evaluating the bonding character in a material. A high value of the bond population indicates a covalent bond, and a low value indicates an ionic bond. Positive and negative values indicate bonding and anti-bonding states, respectively [19,20]. The Mulliken atomic population of Re2P reported in Table 2 shows a significant charge transfer between Re and P, indicating the ionic character of Re and P. The bond population reported in Table 3 shows the strong covalent character of P��Re2II. The chemical bonding in Re2P has predominantly covalent character with mixed covalent- ionic character.
3.3 Elastic properties
Elastic properties are very important for materials because they provide information on interatomic potentials and relate to various fundamental solid state phenomena such as interatomic bonding, equations of state, phonon spectra as well as specific heat, thermal expansion, and Debye temperature [21,22].
Table 1 Calculated lattice parameters and atomic internal coordinate compared with available experimental and theoretic (PDF-PBESOL) data [14] for Re2P

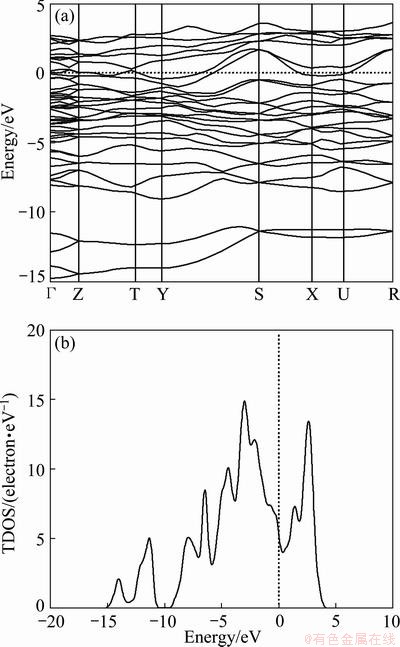
Fig. 2 Band structure calculated by GGA of Re2P along some high-symmetry lines in Brillouin zone (a) and TDOS of Re2P (b) (zero of energy is at Fermi level)
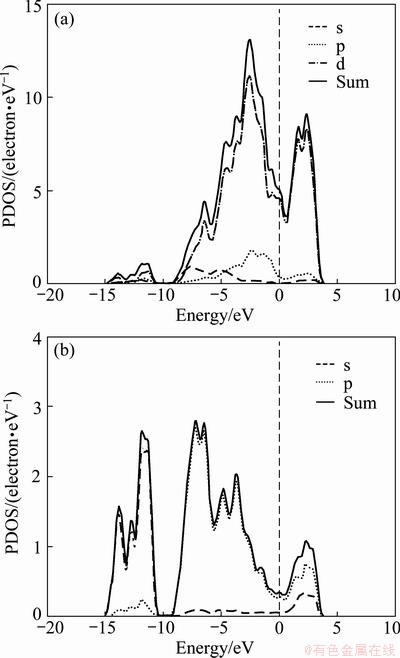
Fig. 3 PDOS of Re (a) and P (b) of Re2P calculated by GGA
Table 2 Mulliken atomic population of Re2P calculated by GGA-USP
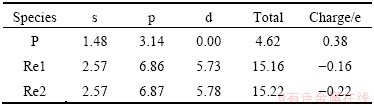
Table 3 Mulliken bond population of Re2P calculated by GGA-USP

Elastic constants are defined by means of a Taylor expansion of the total energy, namely, the derivative of the energy as a function of a lattice strain [15,16]. The orthorhombic phase Re2P crystal has nine independent single crystal elastic constants, C11, C22, C33, C44, C55, C66, C12, C13 and C23 [23]. The Cij value calculated by GGA are presented in Table 4. For the orthorhombic crystal, its mechanical stability requires that its independent elastic constants should satisfy the Born��s stability criteria [23]
 ,
,
 ,
,
 ,
,
 ,
,
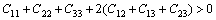 ,
,
 ,
,
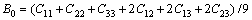 (1)
(1)
From Table 4, it can be seen that these criteria are all satisfied, indicating that Re2P is mechanically stable. The single crystal bulk modulus B is about 294.42 GPa, which is very close to the experimental result [14]. The polycrystal bulk modulus B and shear modulus G are estimated by the Voigt-Reuss-Hill approach in the following forms [23-25]:
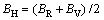 ,
,  (2)
(2)
Elastic modulus E and Poisson ratio v are estimated as follows:
 ,
,
 (3)
(3)
All the calculated results are presented in Table 5. It can be seen that the value of BH/GH ratio for Re2P is 1.62, which is smaller than the critical value (1.75) separating ductile and brittle materials [23-28], indicating that Re2P behaves in a brittle manner.
It is known that the value of the Poisson ratio (��) is the minimal for covalent materials (��=0.1), and increases for ionic systems [29]. In our case, the calculated Poisson ratio is 0.2426, which means a sizable ionic contribution in intra-bonding.
The elastic anisotropy in compressibility (AB) and shear (AG) using the model in Ref. [30] for polycrystalline materials is

 (4)
(4)
A value of zero represents elastic isotropy and a value of 1 is the largest possible anisotropy. The calculated polycrystalline elastic anisotropy in bulk modulus AB is 0.01047 and shear modulus AG is 0.00725. Both the AB and AG are very small. The small anisotropy may minimize cracking propagation during sample preparing and using.
Table 4 Calculated single crystal elastic constants Cij, bulk modulus B and compressibility coefficient �� of Re2P

Table 5 Calculated polycrystalline elastic constants, elastic modulus E, Poisson ratio �� and BH/GH of Re2P by GGA-USP

4 Conclusions
1) The GGA calculated structural parameters of the orthorhombic phase Re2P are in agreement with the available experimental and theoretic data.
2) The electronic band structures and DOS present that Re2P is a metallic material. The DOS and the PDOS calculations show that the DOS near the Fermi level is mainly from the Re-5d state.
3) The chemical bonding analysis shows that Re2P is mainly covalent character with mixed covalent-ionic character. The hard and the ultra-incompressible characteristic of Re2P is ascribed to the mixed covalent-ionic chemical bonding.
4) The elastic constants were calculated and the bulks and shear modulus, elastic modulus, Poisson ratio were derived. All results show that Re2P is mechanically stable and behaves in a brittle manner.
5) The calculated polycrystalline elastic anisotropy in bulk modulus and anisotropy in shear modulus show that orthorhombic phase Re2P is less anisotropy.
References
[1] BRAZHKIN V, DUBROVINSKAIA N, NICOL A. What does harder than diamond mean [J]. Nature Mater, 2004, 3(9): 576-577.
[2] MENG Y, MAO H K, ENG P J. The formation of sp3 bonding in compressed BN [J]. Nature Mater, 2004, 3(2): 111-114.
[3] KURAKEVYCH O O. Superhard phases of simple substances and binary compounds of the B-C-N-O system: From diamond to the latest results [J]. J Superhard Mater, 2009, 31(3): 139-157.
[4] CHUNG H, WEINBERGER M, LEVINE J. Synthesis of ultra-incompressible superhard rhenium diboride at ambient pressure [J]. Science, 2007, 316: 436-439.
[5] GU Q F, KRAUSS G, STEURER W. Transition metal borides: Superhard versus ultra-incompressible [J]. Adv Mater, 2008, 20: 3620-3626.
[6] MOHAMMADI R., LECH A T, XIE M. Tungsten tetraboride, an inexpensive superhard material [J]. Proc Natl Acad Sci USA, 2011, 108(27): 10958-10962.
[7] IVANOVSKII A L. Platinum group metal nitrides and carbides: Synthesis, properties and simulation [J]. Russ Chem Rev, 2009, 78: 303-348.
[8] LI J F, WANG X L, LIU K. Crystal structures, mechanical and electronic properties of tantalum monocarbide and mononitride [J]. J Superhard Mater, 2011, 33(3): 173-178.
[9] ZHENG J C. Superhard hexagonal transition metal and its carbide and nitride: Os, OsC, and OsN [J]. Phys Rev B, 2005, 72: 052105.
[10] ZHAO E J, WANG J P, MENG J. Structural, mechanical and electronic properties of 4d transition metal mononitrides by first-principles [J]. Comput Mater Sci, 2010, 47(4): 1064-1071.
[11] LI Y W, MA Y M. Crystal structure and physical properties of OsN: First-principle calculations [J]. Solid State Commun, 2010, 150(15-16): 759-762.
[12] DU X P, WANG Y X, LO V C. Investigation of tetragonal ReN2 and WN2 with high shear moduli from first-principles calculations [J]. Phys Letts A, 2010, 374(25): 2569-2574.
[13] PENG F, CHEN D, YANG X D. First-principles calculations on elasticity of OsN2 under pressure [J]. Solid State Commun, 2009, 149: 2135-2138.
[14] SCHNEIDER S B, BAUMANN D, SALAMAT A,  Z, LIERMANN H P, SCHWARZ M R, MORGENROTH W, BAYARJARGAL L, FRIEDRICH A, WINKLER B, SCHNICK W. Materials properties of ultra-Incompressible Re2P [J]. Chem Mater, 2012, 24: 3240-3246.
Z, LIERMANN H P, SCHWARZ M R, MORGENROTH W, BAYARJARGAL L, FRIEDRICH A, WINKLER B, SCHNICK W. Materials properties of ultra-Incompressible Re2P [J]. Chem Mater, 2012, 24: 3240-3246.
[15] SEGALL M D, LINDAN PHILIP J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[16] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J, PROBERT M J, REFSON K, PAYNE M C. First principles methods using CASTEP [J]. Zeitschrift fuer Kristallographie, 2005, 220(5-6): 567-570.
[17] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple [J]. Phys Rev Lett, 1996, 77: 3865-3868.
[18] MONKHORST H J, PACK J D. Special points for brillouin-zone integrations [J]. Phys Rev B, 1976, 13: 5188-5192.
[19] HERMET P, GOUMRI-SAID S, KANOUN M B AND HENRARD L. First-principles investigation of the physicalproperties of magnesium nitridoboride [J]. J Phys Chem C, 2009, 113: 4997-5003.
[20] XIA Q L, YI J H, LI Y F, PENG Y D, WANG H Z, ZHOU C S. First-principles investigations of the band structure and optical properties of ��-boron [J]. Solid State Commun, 2010, 150: 605-608.
[21] PONCE C A, CASALI R A, CARAVACA M A. Ab initio study of mechanical and thermo-acoustic properties of tough ceramics: Applications to HfO2 in its cubic and orthorhombic phase [J]. J Phys Condens Mater, 2008, 20: 045213.
[22] BOUHEMADOU A, KHENATA R, CHEGAAR M, MAABED S. First-principles calculations of structural, elastic, electronic and optical properties of the antiperovskite AsNMg3 [J]. Phys Lett A, 2007, 371: 337-343.
[23] NIRANJAN M K. First principles study of structural, electronic and elastic properties of cubic and orthorhombic RhSi [J]. Intermetallics, 2012, 26: 150-156.
[24] SHEIN I R, IVANOVSKII A L. Elastic properties of quaternary oxyarsenide LaOFeAs and LaOFeP as basic phases for new 26-52 K superconducting materials from first principles [J]. Scripta Materialia, 2008, 59: 1099-1102.
[25] SHI Yi-min , YE Shao-long. Chemical bonding and elastic properties of quaternary arsenide oxides YZnAsO and LaZnAsO investigated by first principles [J]. Transactions of Nonferrous Metals Society of China, 2011, 21: 1378-1382.
[26] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proc Phys Soc London A, 1952, 65: 349-355.
[27] XIA Q L, YI J H, LI Y F, PENG Y D, WANG H Z, ZHOU C S. First-principles study of electronic structure and elastic properties of C doping Mg(B1-xCx)2 [J]. Materials Science & Engineering of Powder Metallurgy, 2011, 16(1): 7-12. (in Chinese)
[28] HAINES J , LEGER J M, BOCQUILLON G A. Synthesis and design of superhard materials [J]. Rev Mater Res, 2001, 31: 1-23.
[29] ANDERSON O L. A simplified method for calculating the Debye temperature from elastic constants [J]. J Phys Chem Solids, 1963, 24(7): 909-917.
[30] CHUNG D H, BUESSEM W R. Anisotropy in single crystal refractory compounds [M]. Vol.2. VAHLDIEK F W, MERSOL S A. New York: Plenum Press, 1968: 217.
���˲���ѹ��Re2P�ĵ��ӽṹ����ѧ�����������ʵĵ�һ��ԭ���о�
��һ��1��������2���� ��2
1. ����ʦ����ѧ ��ѧ��������ѧѧԺ�������ܼ����������Ϣ����ʡ�������������ص�ʵ���ң���ɳ 410081��
2. ���ϴ�ѧ ���������ѧԺ����ɳ 410083
ժ Ҫ�����û����ܶȷ�������(DFT)�Ĺ����ݶȽ���(GGA)���о�Co2P���ͽṹ�ļ��˲���ѹ��Re2P�ĵ��ӽṹ����ѧ���͵������ʡ��ܴ��ṹ��ʾRe2PΪ�����Բ��ϣ�̬�ܶȺͷ�̬�ܶȵļ����������������ܼ�������̬�ܶ���Ҫ����Re-5d̬�����ӷ�������Re2P�еĻ�ѧ�������Թ�����Ϊ���Ļ������-��������������õ�Re2P�ľ����������ģ��������ģ���͵����ĵ��Գ������ɴ˵�������ģ���Ͳ��ɱȡ����������Re2P����ѧ�ȶ��ģ��Ҿ���һ���Ĵ��ԡ�
�ؼ��ʣ�Re2P����һ��ԭ�������ӽṹ����ѧ������������
(Edited by Hua YANG)
Foundation item: Project (11271121) supported by the National Natural Science Foundation of China; Project (11JJ2002) supported by the Natural Science Foundation of Hunan Province, China; Project (11K038) supported by Key Laboratory of High Performance Computing and Stochastic Information Processing of Hunan Province, China; Project (2013GK3130) supported by the Scientific and Technological Plan Project of Hunan Province, China
Corresponding author: Yi-fu WANG: +86-731-85881905; E-mail: wyfwyf911@sina.com
DOI: 10.1016/S1003-6326(13)62880-0

